Btrue <- matrix(c( 0.50, -0.10, -0.35, 0.00,
-0.15, 0.60, 0.00, 0.00,
0.30, 0.00, 0.45, -0.30,
0.00, 0.00, 0.25, 0.55), 4, 4, byrow = TRUE)
Atrue <- c(1.20, 0.90, 0.40, 0.30)
Strue <- matrix(c(0.09, 0.03, 0, 0,
0.03, 0.09, 0, 0,
0, 0, 0.04, 0.01,
0, 0, 0.01, 0.04), 4, 4, byrow = TRUE)
p <- 4
sim_mar1 <- function(n, A, B, S, seed, burn = 200) {
set.seed(seed)
p <- length(A)
L <- chol(S)
x <- solve(diag(p) - B, A) # start at the stationary mean
X <- matrix(0, n, p)
for (t in seq_len(burn + n)) {
x <- A + B %*% x + as.vector(rnorm(p) %*% L)
if (t > burn) X[t - burn, ] <- x
}
X
}
X <- sim_mar1(60, Atrue, Btrue, Strue, seed = 4268)Fitting a MAR(1) model to community time series
R
MAR models
time series
community ecology
ecology tutorial
Fit a multivariate autoregressive model to a plankton-like community in base R, and see why the single-species AR(1) shortcut answers a different question.
You have counted four plankton taxa every fortnight for a couple of years. You want the interaction matrix: who suppresses whom, and how hard. The first-order multivariate autoregressive model, MAR(1), is the standard answer to that question in community ecology, and the machinery behind it is about three lines of base R.
This post fits one from scratch. The fitting turns out to be the easy part. The interesting part is what happens when you skip it and fit a separate AR(1) to each species instead, which is what most people reach for first.
The model
Write \(X_t\) for the vector of log abundances of \(p\) species at time \(t\). The MAR(1) process is
\[X_t = A + B\,X_{t-1} + E_t, \qquad E_t \sim \mathrm{MVN}(0, \Sigma).\]
Three objects carry the ecology. \(A\) is a vector of intrinsic growth terms. \(B\) is the \(p \times p\) interaction matrix: \(b_{ij}\) is the effect of species \(j\)’s log abundance on species \(i\)’s per-capita growth rate. \(\Sigma\) is the covariance of the environmental shocks, which is where shared weather, shared sampling error and everything else the model does not name ends up.
The log scale is not decoration. On the log scale a Gompertz competition model is exactly linear, so MAR(1) is either a literal Gompertz community model or a linear approximation to something nonlinear near its equilibrium. Which of those you mean matters later; for now the algebra is the same.
A community to fit
Four taxa, with a mix of strong and weak links and six true zeros. Two of them share a strongly correlated environment later on, so we set up both covariance matrices now.
There is no MASS here: chol(S) plus rnorm draws correlated normals in one line. The process is stationary because the largest eigenvalue modulus of \(B\) is 0.6555, comfortably below one.
Sixty fortnights is a realistic length for a monitoring series, and short enough to be honest about.
series <- data.frame(
time = rep(seq_len(nrow(X)), 4),
logabun = as.vector(X),
species = factor(rep(paste("Species", 1:4), each = nrow(X)))
)
ggplot(series, aes(time, logabun, colour = species)) +
geom_line(linewidth = 0.55) +
scale_colour_manual(values = c(te_forest, te_rust, te_gold, te_sage)) +
labs(title = "A four-species community on the log scale",
subtitle = "Simulated MAR(1) process, 60 time steps",
x = "Time step", y = "Log abundance", colour = NULL) +
theme_te()
Fitting it: three lines
Conditional least squares stacks each time step’s response on the previous step’s state and solves the normal equations once.
mar1_cls <- function(X) {
n <- nrow(X)
Y <- X[-1, , drop = FALSE] # response: steps 2..n
D <- cbind(1, X[-n, , drop = FALSE]) # design: intercept + steps 1..n-1
cf <- solve(crossprod(D), crossprod(D, Y))
E <- Y - D %*% cf
list(A = cf[1, ], B = t(cf[-1, , drop = FALSE]),
Sigma = crossprod(E) / nrow(Y), E = E)
}
fit <- mar1_cls(X)
round(fit$B, 3) [,1] [,2] [,3] [,4]
[1,] 0.580 -0.051 -0.474 -0.044
[2,] -0.133 0.517 -0.180 -0.085
[3,] 0.379 -0.117 0.445 -0.342
[4,] 0.019 -0.016 0.090 0.473That is the whole estimator. The transpose on the way out is the only fiddly bit: cf comes back with predictors in rows and responses in columns, and \(B\) is indexed the other way round.
Standard errors come per cell from the usual least-squares algebra.
mar1_se <- function(X) {
n <- nrow(X); pp <- ncol(X)
Y <- X[-1, , drop = FALSE]
D <- cbind(1, X[-n, , drop = FALSE])
XtXi <- solve(crossprod(D))
q <- nrow(Y); k <- ncol(D)
SE <- matrix(0, pp, pp)
for (i in 1:pp) {
r <- Y[, i] - D %*% (XtXi %*% crossprod(D, Y[, i]))
SE[i, ] <- sqrt(sum(r^2) / (q - k) * diag(XtXi))[-1]
}
SE
}
SE <- mar1_se(X)
round(SE, 3) [,1] [,2] [,3] [,4]
[1,] 0.110 0.093 0.128 0.151
[2,] 0.145 0.123 0.169 0.201
[3,] 0.087 0.074 0.101 0.120
[4,] 0.079 0.066 0.091 0.108The multivariate fit is p separate regressions
Here is the thing that is worth knowing before you write anything about \(B\). Fit species 1’s row with lm, species 2’s row with lm, and so on, ignoring the other species entirely while you do it. Compare.
lmB <- t(sapply(1:p, function(i) coef(lm(X[-1, i] ~ X[-nrow(X), ]))[-1]))
max(abs(fit$B - lmB))[1] 1.376677e-141.377e-14. Machine epsilon. Not close: identical.
The reason is a result older than MAR models. Every equation in the system uses the same design matrix, the lagged state of all species. When the regressors are identical across equations, generalised least squares collapses to equation-by-equation ordinary least squares, and the cross-equation error correlation buys you nothing (Zellner 1962). MAR(1) is a seemingly unrelated regression system in its degenerate case.
Two consequences follow, and they pull in opposite directions.
The comfortable one: the per-cell OLS standard errors above are valid, and you can reason about each row on its own.
The uncomfortable one: the “multivariate” in multivariate autoregressive does no work in estimating \(B\). The multivariate part lives entirely in \(\Sigma\), and \(\Sigma\) is not something the fit uses to find \(B\); it is something the fit reports afterwards. Every claim you make about \(\Sigma\) mattering has to be a claim about something other than the point estimates of \(B\).
Does it recover the truth?
At \(n = 60\) the estimates are noisy but sane. With a long enough series they converge.
Xlong <- sim_mar1(4000, Atrue, Btrue, Strue, seed = 4001)
rbind(true = diag(Btrue), n60 = diag(fit$B), n4000 = diag(mar1_cls(Xlong)$B)) [,1] [,2] [,3] [,4]
true 0.5000000 0.6000000 0.4500000 0.5500000
n60 0.5803138 0.5173230 0.4453603 0.4733782
n4000 0.5156680 0.5958748 0.4688691 0.5476646stat_V <- function(B, S) {
matrix(solve(diag(nrow(B)^2) - kronecker(B, B), as.vector(S)), nrow(B), nrow(B))
}
naive_target <- function(B, S) { V <- stat_V(B, S); diag(B %*% V) / diag(V) }
Scol <- Strue; Scol[1, 2] <- Scol[2, 1] <- 0.087
rec <- data.frame(
true = as.vector(Btrue), est = as.vector(fit$B), se = as.vector(SE),
kind = ifelse(as.vector(diag(4)) == 1, "diagonal", "off-diagonal")
)
p1 <- ggplot(rec, aes(true, est, colour = kind)) +
geom_abline(slope = 1, intercept = 0, colour = te_line, linewidth = 0.7) +
geom_hline(yintercept = 0, colour = te_line, linewidth = 0.3, linetype = 3) +
geom_errorbar(aes(ymin = est - 2 * se, ymax = est + 2 * se), width = 0, linewidth = 0.4) +
geom_point(size = 2) +
scale_colour_manual(values = c(diagonal = te_forest, `off-diagonal` = te_rust)) +
labs(title = "Estimated against true", subtitle = "n = 60, two standard errors",
x = "True coefficient", y = "Estimate", colour = NULL) +
theme_te(11)
nv <- data.frame(
species = rep(paste0("b", 1:4, 1:4), 3),
value = c(diag(Btrue), naive_target(Btrue, Strue), naive_target(Btrue, Scol)),
what = rep(c("true b_ii", "naive target, baseline", "naive target, shared"), each = 4)
)
nv$what <- factor(nv$what, levels = c("true b_ii", "naive target, baseline", "naive target, shared"))
p2 <- ggplot(nv, aes(value, species, colour = what, shape = what)) +
geom_point(size = 2.6) +
scale_colour_manual(values = c(te_ink, te_forest, te_rust)) +
scale_shape_manual(values = c(16, 17, 15)) +
labs(title = "What a per-species AR(1) is aiming at",
subtitle = "Same B, two environments",
x = "Coefficient", y = NULL, colour = NULL, shape = NULL) +
theme_te(11) +
guides(colour = guide_legend(ncol = 1), shape = guide_legend(ncol = 1))
two_panel(p1, p2)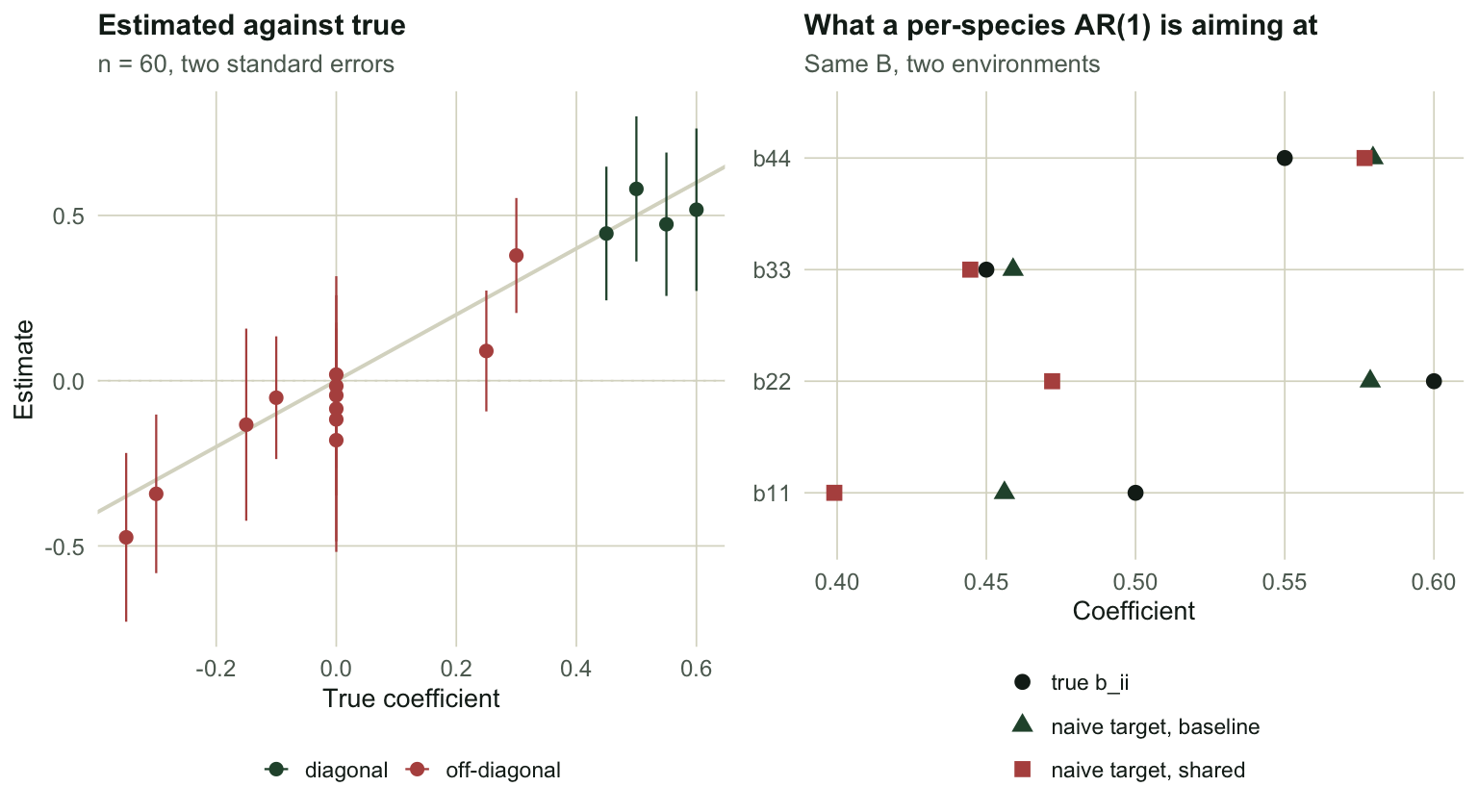
At 4000 steps the diagonal comes back as 0.5157, 0.5959, 0.4689, 0.5477 against a truth of 0.50, 0.60, 0.45, 0.55. The estimator is consistent. Sixty points is simply not four thousand.
The shortcut, and what it actually estimates
Now the part that sends most people wrong. Fitting a separate AR(1) to each species is easy, needs no matrix algebra, and gives you a number per species that looks like \(b_{ii}\). It is tempting to call the difference “bias” and move on.
It is not bias. A per-species AR(1) regresses \(X_{i,t}\) on \(X_{i,t-1}\) alone, so it converges on the population regression coefficient of one on the other, which is \((BV)_{ii}/V_{ii}\) where \(V\) is the stationary covariance of the process. That quantity is a perfectly well-defined thing. It is just not \(b_{ii}\).
rbind(`true b_ii` = diag(Btrue),
`naive aims at` = naive_target(Btrue, Strue)) [,1] [,2] [,3] [,4]
true b_ii 0.5000000 0.6000000 0.4500000 0.5500000
naive aims at 0.4560643 0.5786527 0.4589541 0.5795726The gap is small here. Now change nothing about the ecology. Keep \(B\) exactly as it is, and only make species 1 and 2 share their environment, taking their shock correlation from 0.33 to 0.967.
Va <- stat_V(Btrue, Strue); Vc <- stat_V(Btrue, Scol)
rbind(`true b_ii` = diag(Btrue),
baseline = naive_target(Btrue, Strue),
shared = naive_target(Btrue, Scol)) [,1] [,2] [,3] [,4]
true b_ii 0.5000000 0.6000000 0.4500000 0.5500000
baseline 0.4560643 0.5786527 0.4589541 0.5795726
shared 0.3990504 0.4720548 0.4446178 0.5767508c(cor_baseline = Va[1, 2] / sqrt(Va[1, 1] * Va[2, 2]),
cor_shared = Vc[1, 2] / sqrt(Vc[1, 1] * Vc[2, 2]))cor_baseline cor_shared
0.1495685 0.8454167 The observed correlation between species 1 and 2 goes from 0.150 to 0.845, and the naive answer for species 1 moves from 0.4561 to 0.3991, a shift of 0.057. Species 2 moves by 0.107. The interaction matrix is byte-for-byte the same in both runs. Only \(\Sigma\) changed.
This is the point of the whole exercise. The per-species AR(1) is a consistent estimator of a quantity that mixes \(B\) with \(\Sigma\). If two lakes give you different single-species density dependence, that difference can be entirely environmental correlation, with identical species interactions in both.
Before you write the word “bias” about any shortcut estimator, work out what it is consistent for. Sometimes the shortcut is unbiased for the wrong thing, and that is worse than being biased for the right one, because nothing in the output complains.
The direction of the error is not random
Feed the naive diagonal into the community’s return rate, which is the largest eigenvalue modulus of the interaction matrix, and the errors do not cancel.
c(naive_baseline = max(Mod(eigen(diag(naive_target(Btrue, Strue)))$values)),
naive_shared = max(Mod(eigen(diag(naive_target(Btrue, Scol)))$values)),
truth = max(Mod(eigen(Btrue)$values)))naive_baseline naive_shared truth
0.5795726 0.5767508 0.6555291 True return rate 0.6555; the naive diagonal says 0.5796 in the baseline environment and 0.5768 in the shared one. Both understate persistence, which is to say both make the community look more stable than it is. That is the comfortable direction of error, and it is the direction the shortcut takes in both environments. Checking a MAR(1) model shows a second, larger mechanism pushing the same way.
The honest limit
The gap between the naive answer and \(b_{ii}\) is set by \(\Sigma\), and \(\Sigma\) is exactly what a per-species analysis never sees. So the honest statement is not “the shortcut is off by about 0.05”. It is: the size of the gap depends on the covariance structure, and without fitting the full model you have no way of knowing which case you are in.
That is a good reason to fit the model even when you only care about one species.
Two limits belong to the full model too. MAR(1) assumes linearity on the log scale, and when the underlying dynamics are nonlinear the coefficients recover signs and ranks far more reliably than values (Certain, Barraquand and Gardmark 2018). And it assumes the species you left out do not matter, which no amount of data will tell you.
Where to go next
Interaction strengths from community time series takes the off-diagonal seriously: what the coefficients mean, how badly a 60-point series estimates them, and what shared environments do to identifiability. Community stability from a MAR(1) model turns \(B\) into stability metrics by hand. If you suspect the log-linear assumption is doing too much work, The S-map and state-dependent dynamics is the state-dependent generalisation of the same idea: MAR(1) is the globally linear special case.
For a real analysis, the MARSS package fits this model class with observation error included (Holmes, Ward and Wills 2012), and Hampton et al. (2013) is the ecological review to start from.
References
- Ives A R, Dennis B, Cottingham K L, Carpenter S R (2003) Ecological Monographs 73(2): 301-330.
- Zellner A (1962) Journal of the American Statistical Association 57(298): 348-368. https://doi.org/10.1080/01621459.1962.10480664
- Hampton S E, Holmes E E, Scheef L P, Scheuerell M D, Katz S L, Pendleton D E, Ward E J (2013) Ecology 94(12): 2663-2669. https://doi.org/10.1890/13-0996.1
- Holmes E E, Ward E J, Wills K (2012) The R Journal 4(1): 11-19. https://doi.org/10.32614/RJ-2012-002
- Certain G, Barraquand F, Gardmark A (2018) Methods in Ecology and Evolution 9(9): 1975-1995. https://doi.org/10.1111/2041-210X.13021