---
title: "Model-based unconstrained ordination"
description: "An NMDS axis can track sampling intensity rather than ecology when the mean-variance relationship is strong. A model-based ordination on residuals recovers the real gradient."
date: "2026-08-11 10:00"
categories: [R, community ecology, multivariate, ordination, ecology tutorial]
image: thumbnail.png
image-alt: "Two scatter plots of an ordination axis against a true latent gradient; the model-based points fall close to a line, the distance-based points scatter widely."
---
Non-metric multidimensional scaling is how most community ecologists look at their data: turn the site-by-species table into Bray-Curtis dissimilarities, squeeze them into two dimensions, and read the picture. It is an algorithm for preserving the rank order of dissimilarities, and it works hard at that. What it does not do is account for the statistical properties of counts, and that omission has a cost. Because Bray-Curtis is dominated by the most abundant species, and because abundance and variance travel together, the first axis often ends up tracking how many individuals were caught rather than which species were there.
A model-based ordination starts from the other end. Fit a generalised linear model to the table, with a site effect that absorbs total abundance and a species effect that absorbs how common each species is, then ordinate what is left over: the Dunn-Smyth residuals. Those residuals live on a common scale for every species, so a rare informative species counts as much as a common uninformative one. This post recovers a known gradient with both methods and shows the difference.
```{r setup}
#| echo: false
#| message: false
#| warning: false
library(MASS)
library(ggplot2)
te_ink <- "#16241d"; te_body <- "#2c3a31"; te_green <- "#275139"
te_label <- "#46604a"; te_sage <- "#93a87f"; te_paper <- "#f5f4ee"
te_line <- "#dad9ca"; te_faint <- "#5d6b61"; te_warm <- "#b5534e"; te_gold <- "#c9b458"
theme_te <- function() {
theme_minimal(base_size = 12) +
theme(panel.grid.minor = element_blank(),
panel.grid.major = element_line(colour = "#dad9ca", linewidth = 0.3),
axis.title = element_text(colour = "#2c3a31"),
axis.text = element_text(colour = "#5d6b61"),
plot.title = element_text(colour = "#16241d", face = "bold", size = 13),
plot.subtitle = element_text(colour = "#46604a", size = 10),
strip.text = element_text(colour = "#275139", face = "bold"),
legend.title = element_text(colour = "#2c3a31"),
legend.text = element_text(colour = "#46604a"),
plot.background = element_rect(fill = "white", colour = NA),
panel.background = element_rect(fill = "white", colour = NA))
}
```
## A gradient hidden behind sampling intensity
We simulate 60 sites and 25 species. A single latent gradient `z` drives composition, but the species that respond most strongly to it are the rarer ones; the abundant species that dominate a dissimilarity barely track it. On top of that, sites differ in sampling intensity, a multiplier on every species that has nothing to do with ecology. This is the situation where the two approaches part company.
```{r data}
set.seed(4223)
n <- 60; S <- 25
z <- runif(n, -2, 2) # true latent gradient (unknown to the methods)
b0 <- sort(rnorm(S, mean = log(c(rep(16,6), rep(5,7), rep(1.6,7), rep(0.7,5))), sd = 0.2),
decreasing = TRUE)
lambda <- c(rnorm(6, 0, 0.05), rnorm(7, 0, 0.5), rnorm(7, 0, 1.1), rnorm(5, 0, 1.3))
theta_true <- 1.5
samp <- rnorm(n, 0, 0.6) # sampling intensity, uncorrelated with z
eta <- outer(rep(1,n), b0) + outer(z, lambda) + outer(samp, rep(1,S))
Y <- matrix(rnbinom(n * S, mu = as.vector(exp(eta)), size = theta_true), n, S)
colnames(Y) <- paste0("sp", 1:S)
c(total = sum(Y), min_rowsum = min(rowSums(Y)), max_rowsum = max(rowSums(Y)))
```
Site totals range from `r min(rowSums(Y))` to `r max(rowSums(Y))` individuals, a spread driven by sampling intensity rather than by the gradient we care about.
## Distance-based ordination
We build Bray-Curtis by hand and run NMDS from a classical scaling start, iterating a monotone regression of the configuration distances against the dissimilarity ranks (the Kruskal idea, solved with a Guttman transform).
```{r nmds}
bray <- function(M) { n <- nrow(M); D <- matrix(0, n, n)
for (i in 1:(n-1)) for (j in (i+1):n) {
d <- sum(abs(M[i,] - M[j,])) / sum(M[i,] + M[j,]); D[i,j] <- D[j,i] <- d }; D }
Dbc <- bray(Y)
nmds <- function(D, k = 2, maxit = 300, seed = 1) {
set.seed(seed); n <- nrow(D)
X <- scale(cmdscale(D, k), scale = FALSE)
low <- lower.tri(D); Dl <- D[low]; o <- order(Dl); Sprev <- Inf; Sval <- NA
for (it in 1:maxit) {
dx <- as.matrix(dist(X)); dl <- dx[low]
iso <- isoreg(dl[o])$yf; dhat <- numeric(length(dl)); dhat[o] <- iso
Sval <- sqrt(sum((dl - dhat)^2) / sum(dl^2))
Dh <- matrix(0, n, n); Dh[low] <- dhat; Dh <- Dh + t(Dh)
B <- matrix(0, n, n); nz <- dx > 1e-9; B[nz] <- -Dh[nz] / dx[nz]; diag(B) <- -rowSums(B)
X <- scale((1/n) * B %*% X, scale = FALSE)
if (abs(Sprev - Sval) < 1e-7) break; Sprev <- Sval
}
list(points = X, stress = Sval)
}
mds <- nmds(Dbc, k = 2, seed = 7)
db1 <- mds$points[,1]; db2 <- mds$points[,2]
mds_stress <- mds$stress
round(mds_stress, 3)
```
## Model-based ordination
Now the model. We stack the table into long form, fit a negative binomial GLM with a site term and a species term, and take the Dunn-Smyth residuals: the fitted cumulative probability at each count, jittered across its probability mass and pushed through the normal quantile function. Reshaped back into a matrix, those residuals carry the site-by-species structure that the main effects could not explain, and a singular value decomposition reads off the axes.
```{r modelbased}
#| warning: false
#| message: false
long <- data.frame(count = as.vector(Y),
site = factor(rep(1:n, times = S)),
sp = factor(rep(1:S, each = n)))
fit <- glm.nb(count ~ site + sp, data = long) # site total + species mean, removed
mu_hat <- fitted(fit); th_hat <- fit$theta
ds_resid <- function(y, mu, theta, u) {
a <- pnbinom(y - 1, mu = mu, size = theta)
b <- pnbinom(y, mu = mu, size = theta)
qnorm(a + u * (b - a))
}
set.seed(99)
R <- matrix(ds_resid(long$count, mu_hat, th_hat, runif(nrow(long))), n, S)
sv <- svd(scale(R, center = TRUE, scale = FALSE))
mb1 <- sv$u[,1] * sv$d[1]; mb2 <- sv$u[,2] * sv$d[2]
round(th_hat, 2)
```
## Which axis found the gradient
The test is simple: correlate each ordination's first axis with the true `z`, and also with log total abundance, the nuisance we hoped to be rid of.
```{r recovery}
cor_mb <- abs(cor(mb1, z)); cor_db <- abs(cor(db1, z))
lrs <- log(rowSums(Y))
cor_mb_tot <- abs(cor(mb1, lrs)); cor_db_tot <- abs(cor(db1, lrs))
c(mb_vs_z = round(cor_mb,3), nmds_vs_z = round(cor_db,3),
mb_vs_total = round(cor_mb_tot,3), nmds_vs_total = round(cor_db_tot,3))
```
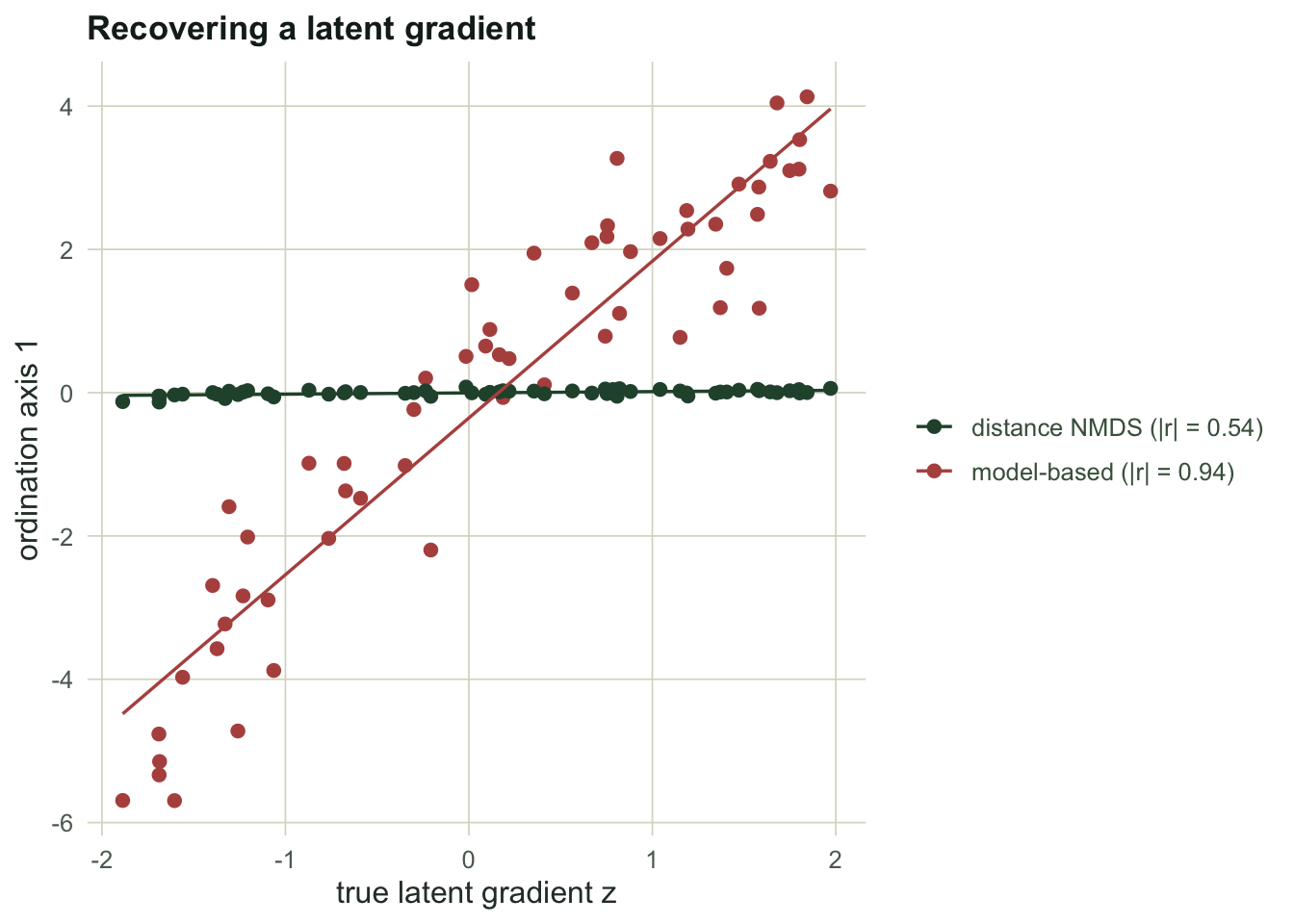
```{r fig-recovery}
#| echo: false
#| fig-alt: "Ordination axis one plotted against the true latent gradient for two methods; the model-based points lie close to a straight line while the distance-based points scatter loosely."
#| fig-cap: "Each point is a site. A tighter line means the ordination axis found the real gradient."
mb1s <- if (cor(mb1, z) < 0) -mb1 else mb1
db1s <- if (cor(db1, z) < 0) -db1 else db1
df <- rbind(data.frame(z = z, axis = mb1s, method = sprintf("model-based (|r| = %.2f)", cor_mb)),
data.frame(z = z, axis = db1s, method = sprintf("distance NMDS (|r| = %.2f)", cor_db)))
ggplot(df, aes(z, axis, colour = method)) +
geom_point(size = 2) + geom_smooth(method = "lm", se = FALSE, linewidth = 0.6) +
scale_colour_manual(values = c(te_green, te_warm)) +
labs(title = "Recovering a latent gradient",
x = "true latent gradient z", y = "ordination axis 1") + theme_te() + theme(legend.title = element_blank())
```
The model-based axis correlates `r round(cor_mb, 2)` with the true gradient; the NMDS axis manages only `r round(cor_db, 2)`. The reason is in the second pair of numbers: the NMDS axis correlates `r round(cor_db_tot, 2)` with log total abundance, so its leading dimension is very nearly the sampling-intensity gradient in disguise. The model-based axis, with the site term already holding total abundance, keeps that correlation down at `r round(cor_mb_tot, 2)`.
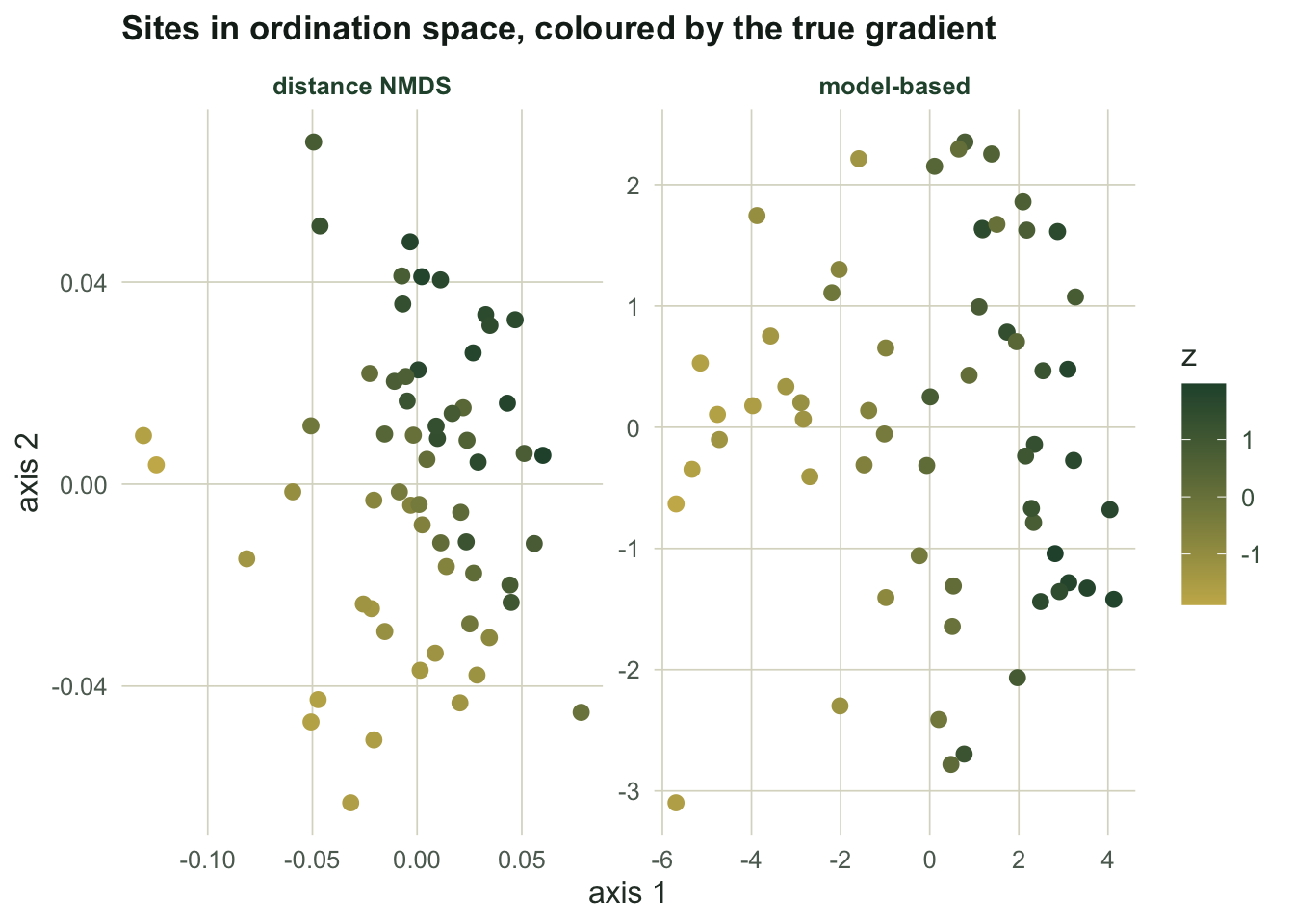
```{r fig-space}
#| echo: false
#| fig-alt: "Two ordination plots of sites, coloured by the true gradient; the model-based panel shows a clean colour sweep across an axis, the distance-based panel shows colours mixed together."
#| fig-cap: "Sites coloured by the true gradient. A clean sweep means the gradient sits on an axis."
op <- rbind(data.frame(a1 = mb1s, a2 = scale(mb2, scale = FALSE)[,1], z = z, panel = "model-based"),
data.frame(a1 = db1s, a2 = db2, z = z, panel = "distance NMDS"))
ggplot(op, aes(a1, a2, colour = z)) + geom_point(size = 2.4) +
facet_wrap(~panel, scales = "free") +
scale_colour_gradient(low = te_gold, high = te_green) +
labs(title = "Sites in ordination space, coloured by the true gradient",
x = "axis 1", y = "axis 2") + theme_te()
```
Note that the NMDS fit is not broken in its own terms: its stress is `r round(mds_stress, 3)`, comfortably in the range usually called a good ordination. It faithfully preserved the dissimilarity ranks. The trouble is that those ranks encode sampling intensity, so a technically good ordination points at the wrong thing. Low stress is a statement about the fit to the dissimilarities, not about ecology.
## Honest limits
The model-based ordination is not free of assumptions, it simply makes them explicit. It assumes a negative binomial mean-variance relationship; if that is wrong the residuals are wrong and the axes drift, which is exactly what the residual diagnostics in the companion post are for. The latent axes are identified only up to rotation and sign, so the numbers on them mean nothing absolute and two runs can look mirrored. Whether to soak up total abundance with a site term is itself a modelling choice: if differences in total abundance are the ecology, removing them is the wrong move. And an ordination of any kind is a description of pattern, not a test of cause. What this example does show is concrete: an NMDS axis can be a sampling-intensity axis wearing an ecological label, and putting species on a common residual scale is what pulls the real gradient back into view.
## References
Hui FKC, Taskinen S, Pledger S, Foster SD, Warton DI 2015. Model-based approaches to unconstrained ordination. Methods in Ecology and Evolution 6:399-411 (10.1111/2041-210X.12236).
Warton DI, Blanchet FG, O'Hara RB, Ovaskainen O, Taskinen S, Walker SC, Hui FKC 2015. So many variables: joint modeling in community ecology. Trends in Ecology and Evolution 30:766-779 (10.1016/j.tree.2015.09.007).
Dunn PK, Smyth GK 1996. Randomized quantile residuals. Journal of Computational and Graphical Statistics 5:236-244 (10.1080/10618600.1996.10474708).
Minchin PR 1987. An evaluation of the relative robustness of techniques for ecological ordination. Vegetatio 69:89-107 (10.1007/BF00038690).
Kruskal JB 1964. Multidimensional scaling by optimizing goodness of fit to a nonmetric hypothesis. Psychometrika 29:1-27 (10.1007/BF02289565).
Legendre P, Legendre L 2012. Numerical Ecology, 3rd edition. Elsevier. ISBN 978-0-444-53868-0.
## Related tutorials
- [Model-based multivariate abundance](../model-based-multivariate-abundance/)
- [Fourth-corner trait-environment analysis](../fourth-corner-trait-environment/)
- [Checking a model-based multivariate model](../checking-a-model-based-multivariate-model/)
- [Common PERMANOVA mistakes](../common-permanova-mistakes/)