library(ggplot2)
set.seed(297)Dispersal limitation: fitting theta and m in R
R
neutral theory
biodiversity
dispersal
likelihood
ecology tutorial
Etienne’s sampling formula in base R. Why fitting Ewens to a dispersal-limited plot underestimates theta by two thirds, and how the immigration parameter fixes it.
The previous post fitted Hubbell’s \(\theta\) with the Ewens sampling formula, and the formula assumes your plot is a random draw from the metacommunity. Every individual in your quadrat arrived independently from the whole species pool.
No real plot works like that. A seed that lands under its parent is not a random draw from anything. That gap has a name in the theory, dispersal limitation, and a parameter, the immigration probability \(m\). This post fits \(\theta\) and \(m\) together, and measures what happens if you do not.
Setup
The local community
Hubbell’s local community is a second urn sitting downstream of the first. Fill a plot of \(J\) individuals one at a time. Each new individual is an immigrant from the metacommunity with probability \(m\), and otherwise a copy of an individual already in the plot. Immigrants carry metacommunity species labels, so the metacommunity urn from the previous post supplies them.
It is more convenient to work with the fundamental dispersal number \(I\), which converts to \(m\) by \(I = m(J-1)/(1-m)\). The reason for the reparameterisation is that \(I\) plays exactly the role \(\theta\) plays one level up: at step \(k\), the next individual is an immigrant with probability \(I/(I+k)\).
sim_etienne <- function(theta, I, J) {
loc <- integer(J); anc <- integer(0); nsp <- 0L
for (k in 0:(J - 1)) {
if (runif(1) < I / (I + k)) { # immigracios esemeny
A <- length(anc)
if (A == 0 || runif(1) < theta / (theta + A)) { # a bevandorlo uj faj
nsp <- nsp + 1L; s <- nsp
} else { # egy korabbi bevandorlo faja
s <- anc[sample.int(A, 1)]
}
anc <- c(anc, s); loc[k + 1] <- s
} else { # lokalis szaporodas
loc[k + 1] <- loc[sample.int(k, 1)]
}
}
as.integer(table(loc))
}
m_to_I <- function(m, J) m * (J - 1) / (1 - m)
I_to_m <- function(I, J) I / (I + J - 1)The nesting is what makes the model work: the plot samples \(A\) immigration events from the metacommunity, and those \(A\) ancestors are an Ewens sample. Since the immigration urn has the same structure as the Hoppe urn, the expected number of immigration events reuses the digamma formula from the previous post.
ES <- function(theta, J) theta * (digamma(theta + J) - digamma(theta))
theta_true <- 50; m_true <- 0.05; J <- 1000
I_true <- m_to_I(m_true, J)
EA <- ES(I_true, J) # varhato immigracios esemenyszam
c(I = I_true, E_immigration_events = EA, E_S_from_those_events = ES(theta_true, EA),
E_S_if_m_were_1 = ES(theta_true, J)) I E_immigration_events E_S_from_those_events
52.57895 158.03903 71.66654
E_S_if_m_were_1
152.70398 The last two numbers are the whole problem. A thousand individuals drawn straight from the metacommunity would hold about 153 species. The same thousand individuals with five percent immigration hold about 72, because they descend from only about 158 independent arrivals. Dispersal limitation does not just reshape the abundance curve, it destroys most of the richness.
What Ewens does with such a plot
The previous post showed that the Ewens fit reads \(S\) and \(J\) and nothing else. Feed it a dispersal-limited plot and it sees a poor, apparently species-starved sample, and reports the only \(\theta\) that could produce it under \(m = 1\).
theta_mle <- function(S, J) {
if (S <= 1 || S >= J) return(NA_real_)
uniroot(function(th) ES(th, J) - S, c(1e-8, 1e6), tol = 1e-10)$root
}
d1 <- sort(sim_etienne(theta_true, I_true, J), decreasing = TRUE)
c(S_observed = length(d1), J = sum(d1), theta_naive = theta_mle(length(d1), J), theta_true = theta_true) S_observed J theta_naive theta_true
69.00000 1000.00000 16.66303 50.00000 One sample proves nothing, so we repeat it later. First we need the correction.
Etienne’s sampling formula
Etienne (2005) marginalised the unobserved number of immigration events out of the likelihood, the same move that makes an N-mixture or an occupancy model work. Conditional on \(A\) ancestors, the plot is a known combinatorial object; sum over all \(A\) from \(S\) to \(J\):
\[P(D \mid \theta, I) = \frac{J!}{\prod_i n_i \prod_j \Phi_j!} \cdot \frac{1}{(I)_J} \sum_{A=S}^{J} K(D, A) \, \frac{\theta^S}{(\theta)_A} \, I^A\]
The weights \(K(D,A)\) are the awkward part. They count the ways the observed abundances can descend from \(A\) ancestors, they depend only on the data, and they are built from unsigned Stirling numbers of the first kind. Because they do not involve \(\theta\) or \(I\), you compute them once and reuse them at every step of the optimiser.
log_stirling1 <- function(N) { # log |s(n,k)|, n = 0..N
L <- matrix(-Inf, N + 1, N + 1); L[1, 1] <- 0
for (n in 0:(N - 1)) for (k in 0:(n + 1)) {
a <- if (k >= 1) L[n + 1, k] else -Inf
b <- if (n >= 1) log(n) + L[n + 1, k + 1] else -Inf
mx <- max(a, b)
L[n + 2, k + 1] <- if (is.infinite(mx)) -Inf else mx + log(exp(a - mx) + exp(b - mx))
}
L
}
LS5 <- log_stirling1(5)
round(exp(LS5[6, 2:6])) # s(5,k), k = 1..5[1] 24 50 35 10 1Those are 24, 50, 35, 10, 1, and they sum to 120, which is \(5!\), as unsigned Stirling numbers of the first kind must.
A trap worth knowing about
\(K(D, \cdot)\) is a convolution of per-species vectors, and R ships a fast convolution in stats::convolve. Using it here is a mistake. The per-species terms span twenty orders of magnitude, and convolve works through the FFT, whose round-off is relative to the largest term in the transform. The small entries come back as noise, and some come back negative. Write the direct version instead; it is a few lines and it is fast enough.
conv_direct <- function(a, b) {
if (length(a) > length(b)) { tmp <- a; a <- b; b <- tmp } # a rovidebbre hurkolj
r <- numeric(length(a) + length(b) - 1)
for (i in seq_along(a)) r[i + seq_along(b) - 1] <- r[i + seq_along(b) - 1] + a[i] * b
r
}
logK <- function(n, LS) {
S <- length(n); Jn <- sum(n)
cur <- 1; off <- 0
for (i in seq_len(S)) {
a <- 1:n[i]
lv <- LS[n[i] + 1, a + 1] + lgamma(a) - lgamma(n[i]) # log s(n_i,a) + log(a-1)! - log(n_i-1)!
M <- max(lv); off <- off + M
cur <- conv_direct(cur, exp(lv - M))
m2 <- max(cur); cur <- cur / m2; off <- off + log(m2) # lepesenkent ujranormalva
}
out <- rep(-Inf, Jn + 1) # index: A + 1
out[(S:Jn) + 1] <- log(pmax(cur[(S:Jn) - S + 1], 0)) + off
out
}The identity that catches the FFT version is exact and costs nothing to check: \(K(D, S) = K(D, J) = 1\) for any \(D\), because there is exactly one way for the sample to descend from \(S\) ancestors (one per species) and exactly one way for it to descend from \(J\) (one per individual).
LK1 <- logK(d1, log_stirling1(max(d1)))
c(K_at_S = exp(LK1[length(d1) + 1]), K_at_J = exp(LK1[J + 1]))K_at_S K_at_J
1 1 Now the likelihood itself.
lpoch <- function(x, n) if (n == 0) 0 else sum(log(x + 0:(n - 1)))
sci_tex <- function(x, d = 1) { # olvashato kitevos alak a prozaba
e <- floor(log10(abs(x))); sprintf("$%.*f \\times 10^{%d}$", d, x / 10^e, e)
}
etienne_loglik <- function(n, theta, I, LK) {
Jn <- sum(n); S <- length(n)
const <- lfactorial(Jn) - sum(log(n)) - sum(lfactorial(as.numeric(table(n))))
A <- S:Jn
lpt <- cumsum(log(theta + 0:(Jn - 1))) # (theta)_A minden A-ra, egyszerre
tt <- LK[A + 1] + S * log(theta) - lpt[A] + A * log(I)
mx <- max(tt)
const + mx + log(sum(exp(tt - mx))) - lpoch(I, Jn) # log-sum-exp
}
fit_etienne <- function(n) {
Jn <- sum(n); LK <- logK(n, log_stirling1(max(n))) # EGYSZER, az optimalizalas ELOTT
f <- optim(c(log(20), log(20)),
function(p) -etienne_loglik(n, exp(p[1]), exp(p[2]), LK),
method = "Nelder-Mead", control = list(reltol = 1e-12, maxit = 3000))
list(theta = exp(f$par[1]), I = exp(f$par[2]), m = I_to_m(exp(f$par[2]), Jn), logL = -f$value)
}Two checks before it is allowed near data. The formula must be a probability distribution, so enumerate all abundance vectors for a small \(J\) and add them up. And as \(I\) grows, every individual becomes an immigrant, \(m\) approaches one, and the formula must collapse back to Ewens.
parts <- function(Jn, mx = Jn) {
if (Jn == 0) return(list(integer(0)))
out <- list(); for (k in min(Jn, mx):1) for (p in parts(Jn - k, k)) out[[length(out) + 1]] <- c(k, p)
out
}
P6 <- parts(6)
tot6 <- sapply(list(c(3, 8), c(20, 2)), function(pr)
sum(sapply(P6, function(n) exp(etienne_loglik(n, pr[1], pr[2], logK(n, log_stirling1(max(n))))))))
esf_loglik <- function(n, theta) {
const <- lfactorial(sum(n)) - sum(log(n)) - sum(lfactorial(as.numeric(table(n))))
const + length(n) * log(theta) - lpoch(theta, sum(n))
}
dsm <- c(4, 2, 2, 1, 1)
lim <- sapply(10^(3:9), function(Ibig)
etienne_loglik(dsm, 6, Ibig, logK(dsm, log_stirling1(max(dsm)))) - esf_loglik(dsm, 6))
data.frame(I = 10^(3:9), gap_vs_Ewens = signif(lim, 3), gap_times_I = signif(lim * 10^(3:9), 3)) I gap_vs_Ewens gap_times_I
1 1e+03 1.48e-02 14.8
2 1e+04 1.50e-03 15.0
3 1e+05 1.50e-04 15.0
4 1e+06 1.50e-05 15.0
5 1e+07 1.50e-06 15.0
6 1e+08 1.50e-07 15.0
7 1e+09 1.50e-08 15.0Both sums are 1. And the gap to the Ewens likelihood shrinks exactly in proportion to \(1/I\): multiplying the gap by \(I\) gives a constant, 15.0, holding from \(I = 10^4\) up to \(I = 10^9\). That is the behaviour a correct nesting must show, and it is the kind of check an FFT-based logK fails loudly.
The measurement
Simulate three hundred plots at a known \(\theta\) and \(m\), fit each one both ways, and compare.
B <- 300
mc <- t(replicate(B, {
d <- sort(sim_etienne(theta_true, I_true, J), decreasing = TRUE)
f <- fit_etienne(d)
c(theta_naive = theta_mle(length(d), J), theta_et = f$theta, m_et = f$m, S = length(d))
}))
q <- function(x) c(median = median(x), lo = unname(quantile(x, 0.05)), hi = unname(quantile(x, 0.95)))
round(rbind(theta_naive = q(mc[, "theta_naive"]), theta_etienne = q(mc[, "theta_et"]),
m_etienne = q(mc[, "m_et"]), S_observed = q(mc[, "S"])), 4) median lo hi
theta_naive 17.3065 14.1655 20.9800
theta_etienne 54.3841 28.1915 180.9956
m_etienne 0.0461 0.0217 0.1407
S_observed 71.0000 61.0000 82.0000The naive Ewens fit reports a median \(\theta\) of 17.3 against a true 50, an error of -65%. It is not noisy, it is wrong in a fixed direction: 100% of the three hundred estimates land below the truth. Etienne’s formula recovers 54.4 for \(\theta\) and 0.0461 for \(m\), against 50 and 0.05.
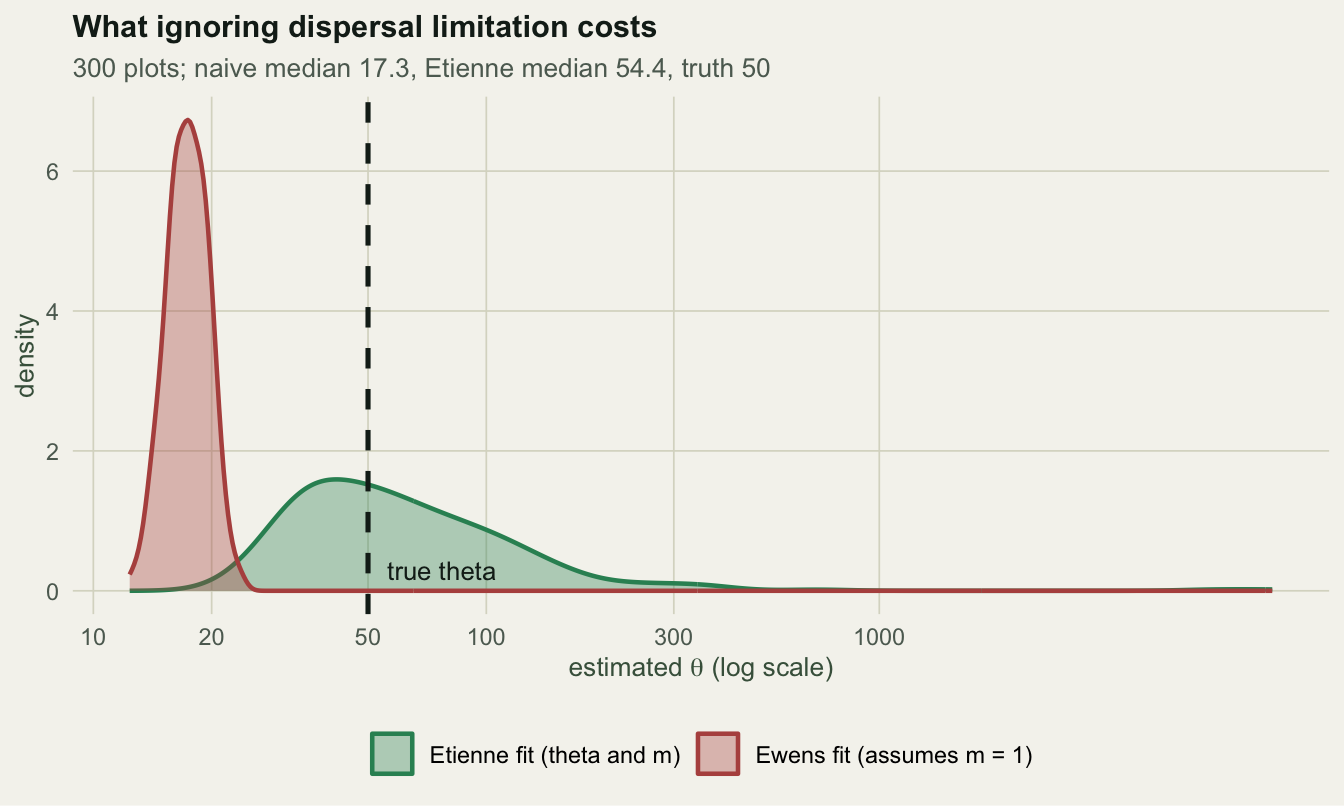
The abundances now matter, and one of them refuses to answer
One consequence is worth checking directly. Under Ewens, the two samples from the previous post gave identical fits because both had \(S = 20\) and \(J = 200\). Under Etienne they do not.
even <- rep(10L, 20)
uneven <- c(100L, 48L, 24L, 12L, rep(1L, 16))
fe <- fit_etienne(sort(even, decreasing = TRUE))
fu <- fit_etienne(sort(uneven, decreasing = TRUE))
rbind(even = c(theta = fe$theta, I = fe$I, m = fe$m, logL = fe$logL),
uneven = c(theta = fu$theta, I = fu$I, m = fu$m, logL = fu$logL)) theta I m logL
even 1.031602e+01 27.576972 0.1217113 -68.77381
uneven 3.958450e+13 5.343543 0.0261498 -30.65623The even sample gets a sensible interior fit. The uneven one sends \(\theta\) to \(4.0 \times 10^{13}\), which looks like an optimiser that lost its footing. It is not. It is an exact limit of the formula, and the formula is telling us something.
Look at the sum over \(A\). Each term carries \(\theta^S / (\theta)_A\), and for large \(\theta\) the rising factorial behaves like \(\theta^A\), so the term scales as \(\theta^{S-A}\). Every \(A\) above \(S\) is crushed. Only \(A = S\) survives, where \(K(D,S) = 1\), and what is left is
\[\lim_{\theta \to \infty} P(D \mid \theta, I) = \frac{J!}{\prod_i n_i \prod_j \Phi_j!} \cdot \frac{I^S}{(I)_J}\]
which is the Ewens sampling formula with \(I\) in place of \(\theta\). An infinitely diverse metacommunity makes every immigrant a new species, the two levels collapse into one, and the immigration number takes over the role the biodiversity number played.
theta_grid <- 10^(2:12)
gap <- sapply(theta_grid, function(th) etienne_loglik(uneven, th, 5, LK = logK(uneven, log_stirling1(max(uneven)))) -
esf_loglik(uneven, 5))
th_ew <- theta_mle(length(uneven), sum(uneven))
c(gap_at_theta_1e12 = gap[11], I_hat_uneven = fu$I, theta_Ewens_uneven = th_ew,
difference = abs(fu$I - th_ew), logL_gap = abs(fu$logL - esf_loglik(uneven, th_ew))) gap_at_theta_1e12 I_hat_uneven theta_Ewens_uneven difference
-1.080025e-10 5.343543e+00 5.343537e+00 5.493554e-06
logL_gap
9.549694e-12 The check is exact twice over. The Etienne likelihood at \(\theta = 10^{12}\) sits \(1.1 \times 10^{-10}\) from the Ewens likelihood at \(I\), and the fitted \(\hat I\) for the uneven sample equals its Ewens \(\hat\theta\) to \(5.5 \times 10^{-6}\), with log-likelihoods agreeing to \(9.5 \times 10^{-12}\). The two fits are the same fit.
So the abundance shape did get a vote, and on the uneven sample the vote was a refusal. The profile likelihood makes the refusal visible: it climbs, flattens, and never turns back.
LKu <- logK(uneven, log_stirling1(max(uneven)))
prof <- sapply(10^(1:8), function(th)
-optimize(function(li) -etienne_loglik(uneven, th, exp(li), LK = LKu), c(log(0.2), log(4000)))$objective)
data.frame(theta = 10^(1:8), profile_logL = round(prof, 3)) theta profile_logL
1 1e+01 -34.026
2 1e+02 -31.629
3 1e+03 -30.758
4 1e+04 -30.666
5 1e+05 -30.657
6 1e+06 -30.656
7 1e+07 -30.656
8 1e+08 -30.656Read that as an ecological statement rather than a numerical one. The best explanation of a plot with one species holding half the individuals and sixteen singletons is: twenty species arrived as twenty separate immigrants, and local reproduction did everything after that. Nothing in the sample bounds how diverse the source pool was, so nothing bounds \(\theta\). The even sample, by contrast, does bound it, and beats the one-parameter Ewens fit by 5.5 log units.
The boundary case is not rare, and the checking post measures how often it happens on data the model generated itself.
Honest limits
Theta and m trade off. Across the three hundred simulated plots, the two estimates are negatively correlated on the log scale, -0.45. The likelihood has a ridge: a plot that looks species-poor can be explained by a small metacommunity or by a plot that rarely receives immigrants, and the data separate the two weakly. The ninety-fifth percentile of \(\hat\theta\) sits at 181, four times the truth, on data generated by the model itself.
Warning in geom_point(aes(x = f1$theta, y = f1$m), inherit.aes = FALSE, : All aesthetics have length 1, but the data has 3600 rows.
ℹ Please consider using `annotate()` or provide this layer with data containing
a single row.Warning in geom_point(aes(x = theta_true, y = m_true), inherit.aes = FALSE, : All aesthetics have length 1, but the data has 3600 rows.
ℹ Please consider using `annotate()` or provide this layer with data containing
a single row.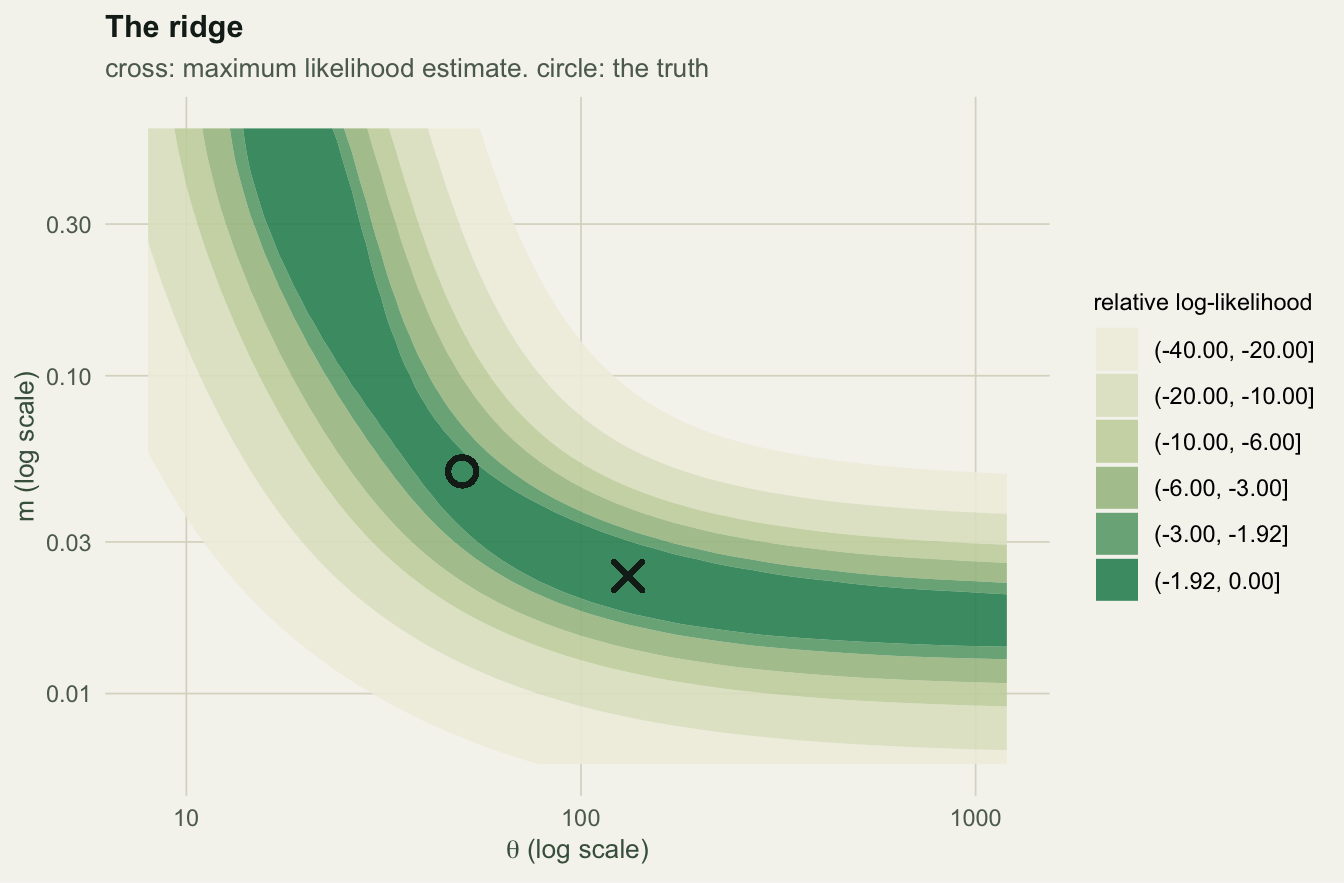
The correction is not a licence. Recovering \(\theta\) and \(m\) on data the model generated is the minimum a fitting routine must do, not evidence that a real plot is neutral. Etienne, Alonso and McKane (2007) showed that several of the theory’s structural assumptions, the zero-sum constraint among them, can be relaxed without changing the sampling formula at all, which means fitting it well tests fewer of them than the derivation suggests.
Two parameters, one snapshot. \(m\) is a per-birth immigration probability, estimated from a single census with no dispersal data in it. It absorbs anything that makes a plot more aggregated than a random metacommunity draw: clonal growth, seed shadow, a patch of favourable soil, or a competitor holding a site. Only the first of those is dispersal.
Where to go next
Fitting the model is the easy half. The next two posts do the harder half: a test of whether the abundance shape is neutral at all, and a set of checks on the fit itself.
References
- Etienne 2005 Ecology Letters 8(3):253-260 (10.1111/j.1461-0248.2004.00717.x)
- Etienne & Alonso 2005 Ecology Letters 8(11):1147-1156 (10.1111/j.1461-0248.2005.00817.x)
- Etienne, Alonso & McKane 2007 Journal of Theoretical Biology 248(3):522-536 (10.1016/j.jtbi.2007.06.010)
- Alonso & McKane 2004 Ecology Letters 7(10):901-910 (10.1111/j.1461-0248.2004.00640.x)
- Hubbell 2001 The Unified Neutral Theory of Biodiversity and Biogeography, ISBN 978-0-691-02128-7
- Rosindell, Hubbell & Etienne 2011 Trends in Ecology and Evolution 26(7):340-348 (10.1016/j.tree.2011.03.024)