library(ggplot2)
set.seed(299)Checking a neutral model fit
R
neutral theory
model checking
likelihood
ecology tutorial
Three checks on a fitted neutral model in R: interval calibration, out-of-sample richness prediction on nested subsamples, and what stays unidentifiable.
The previous two posts fit Hubbell’s model and tested its abundance prediction. This one asks what the fit is worth once you have it. Three checks, in the order that finds problems fastest: does the interval mean what it says, does the fitted model predict anything it was not shown, and what does the likelihood refuse to identify no matter how much data you collect.
The pattern generalises past neutral theory. The checks are cheap, they are all Monte Carlo, and each one has to be run on data where it should fail as well as data where it should pass. A check that cannot fail is not a check.
Setup
The machinery is from the previous two posts, compressed. The Ewens side needs one root find; the Etienne side needs Stirling numbers and a convolution.
ES <- function(theta, J) theta * (digamma(theta + J) - digamma(theta))
theta_mle <- function(S, J) {
if (S <= 1 || S >= J) return(NA_real_)
uniroot(function(th) ES(th, J) - S, c(1e-8, 1e6), tol = 1e-10)$root
}
lpoch <- function(x, n) if (n == 0) 0 else sum(log(x + 0:(n - 1)))
log_stirling1 <- function(N) {
L <- matrix(-Inf, N + 1, N + 1); L[1, 1] <- 0
for (n in 0:(N - 1)) for (k in 0:(n + 1)) {
a <- if (k >= 1) L[n + 1, k] else -Inf
b <- if (n >= 1) log(n) + L[n + 1, k + 1] else -Inf
mx <- max(a, b)
L[n + 2, k + 1] <- if (is.infinite(mx)) -Inf else mx + log(exp(a - mx) + exp(b - mx))
}
L
}
conv_direct <- function(a, b) {
if (length(a) > length(b)) { tmp <- a; a <- b; b <- tmp }
r <- numeric(length(a) + length(b) - 1)
for (i in seq_along(a)) r[i + seq_along(b) - 1] <- r[i + seq_along(b) - 1] + a[i] * b
r
}
logK <- function(n, LS) {
S <- length(n); Jn <- sum(n); cur <- 1; off <- 0
for (i in seq_len(S)) {
a <- 1:n[i]
lv <- LS[n[i] + 1, a + 1] + lgamma(a) - lgamma(n[i])
M <- max(lv); off <- off + M
cur <- conv_direct(cur, exp(lv - M))
m2 <- max(cur); cur <- cur / m2; off <- off + log(m2)
}
out <- rep(-Inf, Jn + 1); out[(S:Jn) + 1] <- log(pmax(cur[(S:Jn) - S + 1], 0)) + off; out
}
etienne_loglik <- function(n, theta, I, LK) {
Jn <- sum(n); S <- length(n)
const <- lfactorial(Jn) - sum(log(n)) - sum(lfactorial(as.numeric(table(n))))
A <- S:Jn; lpt <- cumsum(log(theta + 0:(Jn - 1)))
tt <- LK[A + 1] + S * log(theta) - lpt[A] + A * log(I)
mx <- max(tt); const + mx + log(sum(exp(tt - mx))) - lpoch(I, Jn)
}
sim_etienne <- function(theta, I, J) {
loc <- integer(J); anc <- integer(0); ns <- 0L
for (k in 0:(J - 1)) {
if (runif(1) < I / (I + k)) {
A <- length(anc)
if (A == 0 || runif(1) < theta / (theta + A)) { ns <- ns + 1L; s <- ns } else s <- anc[sample.int(A, 1)]
anc <- c(anc, s); loc[k + 1] <- s
} else loc[k + 1] <- loc[sample.int(k, 1)]
}
as.integer(table(loc))
}
m_to_I <- function(m, J) m * (J - 1) / (1 - m)Check one: does the interval mean what it says
A confidence interval that covers the truth 95% of the time is doing its job. The only way to know is to generate data from the model at a known parameter and count. Fit each simulated plot, then invert the profile likelihood.
fit2 <- function(n, LK) {
Jn <- sum(n)
f <- optim(c(log(20), log(20)), function(p) -etienne_loglik(n, exp(p[1]), exp(p[2]), LK),
method = "Nelder-Mead", control = list(reltol = 1e-12, maxit = 3000))
list(theta = exp(f$par[1]), I = exp(f$par[2]), m = exp(f$par[2]) / (exp(f$par[2]) + Jn - 1), logL = -f$value)
}
prof <- function(n, LK, th) -optimize(function(li) -etienne_loglik(n, th, exp(li), LK), c(log(0.05), log(1e6)))$objective
th0 <- 50; m0 <- 0.05; Jloc <- 1000; I0 <- m_to_I(m0, Jloc); Bci <- 200
CI <- t(sapply(1:Bci, function(b) {
d <- sort(sim_etienne(th0, I0, Jloc), decreasing = TRUE)
LK <- logK(d, log_stirling1(max(d))); f <- fit2(d, LK)
cut <- f$logL - qchisq(0.95, 1) / 2
g <- function(x) prof(d, LK, x) - cut
c(theta = f$theta, m = f$m,
lo = tryCatch(uniroot(g, c(1, f$theta))$root, error = function(e) 1),
hi = tryCatch(uniroot(g, c(f$theta, 1e6))$root, error = function(e) Inf))
}))
cov <- mean(CI[, "lo"] <= th0 & CI[, "hi"] >= th0)
unb <- mean(CI[, "hi"] > 1e5)
wid <- median(log10(pmin(CI[, "hi"], 1e6) / CI[, "lo"]))
rho <- cor(log(CI[, "theta"]), log(CI[, "m"]))
round(c(median_theta = median(CI[, "theta"]), true_theta = th0,
median_m = median(CI[, "m"]), true_m = m0,
coverage = cov, unbounded_above = unb, median_width_orders = wid, cor_log_theta_log_m = rho), 4) median_theta true_theta median_m true_m
51.3731 50.0000 0.0507 0.0500
coverage unbounded_above median_width_orders cor_log_theta_log_m
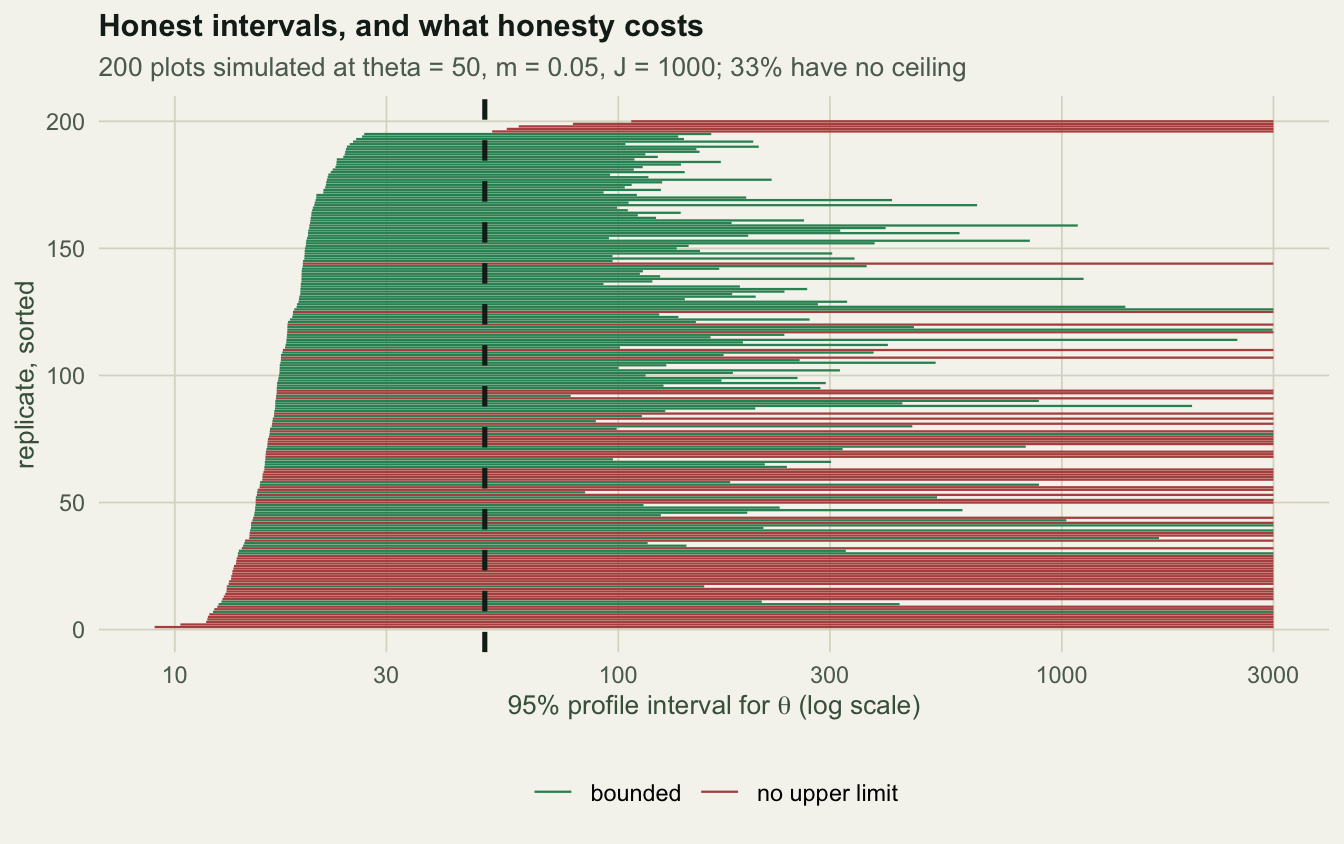
0.9750 0.3300 1.2794 -0.3460 The point estimates are close to the truth: median \(\hat\theta\) = 51.4 against 50, median \(\hat m\) = 0.0507 against 0.05. Coverage is 0.975 against a nominal 0.95, slightly conservative. The fit passes.
Now read the same numbers again. The interval covers 98% of the time partly because in 33% of replicates it has no upper limit at all: the profile climbs to a plateau and never comes back down, exactly the boundary case the dispersal post hit on a hand-built sample. The typical interval spans 1.3 orders of magnitude. And \(\hat\theta\) and \(\hat m\) are correlated at -0.35 on the log scale, which is the ridge.
This is what a calibration check is for. The interval is honest, and its honesty is the bad news. On data generated by the model, with a thousand individuals, a third of the time the data cannot put a ceiling on the metacommunity diversity. A published point estimate of \(\theta\) without an interval is close to content free.
Check two: does the fit predict anything
A model fitted to a sample and then judged on that same sample is grading its own homework. The Ewens formula gives a way out that costs nothing, because it predicts richness at every sample size from a single \(\theta\):
\[\mathrm{E}[S_J] = \theta\,\bigl(\psi(\theta + J) - \psi(\theta)\bigr)\]
So fit \(\theta\) to a nested subsample of 250 individuals, predict the richness of the full 8000, and compare with what is there. Kingman’s consistency guarantees that a subsample of an Ewens sample is an Ewens sample with the same \(\theta\), so under the null this must work.
sim_esf_ind <- function(theta, J) { # egyed-cimkek, veletlen sorrendben
sp <- integer(J); sp[1] <- 1L; ns <- 1L
for (k in 1:(J - 1)) {
if (runif(1) < theta / (theta + k)) { ns <- ns + 1L; sp[k + 1] <- ns } else sp[k + 1] <- sp[sample.int(k, 1)]
}
sp
}
sugihara <- function(Sp, f = 0.75) {
seg <- 1
while (length(seg) < Sp) {
i <- sample.int(length(seg), 1); s <- seg[i]
g <- if (runif(1) < 0.5) f else 1 - f
seg <- c(seg[-i], s * g, s * (1 - g))
}
seg / sum(seg)
}
Js <- c(250, 500, 1000, 2000, 4000, 8000); Jmax <- 8000; Bd <- 150
nested <- function(gen) {
R <- t(replicate(Bd, {
ind <- gen()
c(sapply(Js, function(j) theta_mle(length(unique(ind[1:j])), j)),
sapply(Js, function(j) length(unique(ind[1:j]))))
}))
TH <- R[, 1:6]; SV <- R[, 7:12]
pred <- ES(TH[, 1], Jmax) # a J=250-bol illesztett theta jelzese
list(theta = apply(TH, 2, median), S = apply(SV, 2, median),
ratio = median(TH[, 6] / TH[, 1]),
pred = median(pred), obs = median(SV[, 6]),
hit = mean(abs(SV[, 6] / pred - 1) < 0.10))
}
gens <- list(
"neutral, theta = 15" = function() sim_esf_ind(15, Jmax),
"broken stick, pool 60" = function() sample.int(60, Jmax, TRUE, prob = diff(sort(c(0, runif(59), 1)))),
"Sugihara, pool 80" = function() sample.int(80, Jmax, TRUE, prob = sugihara(80)),
"Sugihara, pool 200" = function() sample.int(200, Jmax, TRUE, prob = sugihara(200)))
NS <- lapply(gens, nested)
data.frame(model = names(gens),
theta_at_250 = round(sapply(NS, function(z) z$theta[1]), 1),
theta_at_8000 = round(sapply(NS, function(z) z$theta[6]), 1),
ratio = round(sapply(NS, function(z) z$ratio), 2),
predicted_S = round(sapply(NS, function(z) z$pred)),
observed_S = round(sapply(NS, function(z) z$obs)),
error = sprintf("%+.0f%%", 100 * (sapply(NS, function(z) z$pred / z$obs) - 1)),
within_10pct = sprintf("%.0f%%", 100 * sapply(NS, function(z) z$hit)),
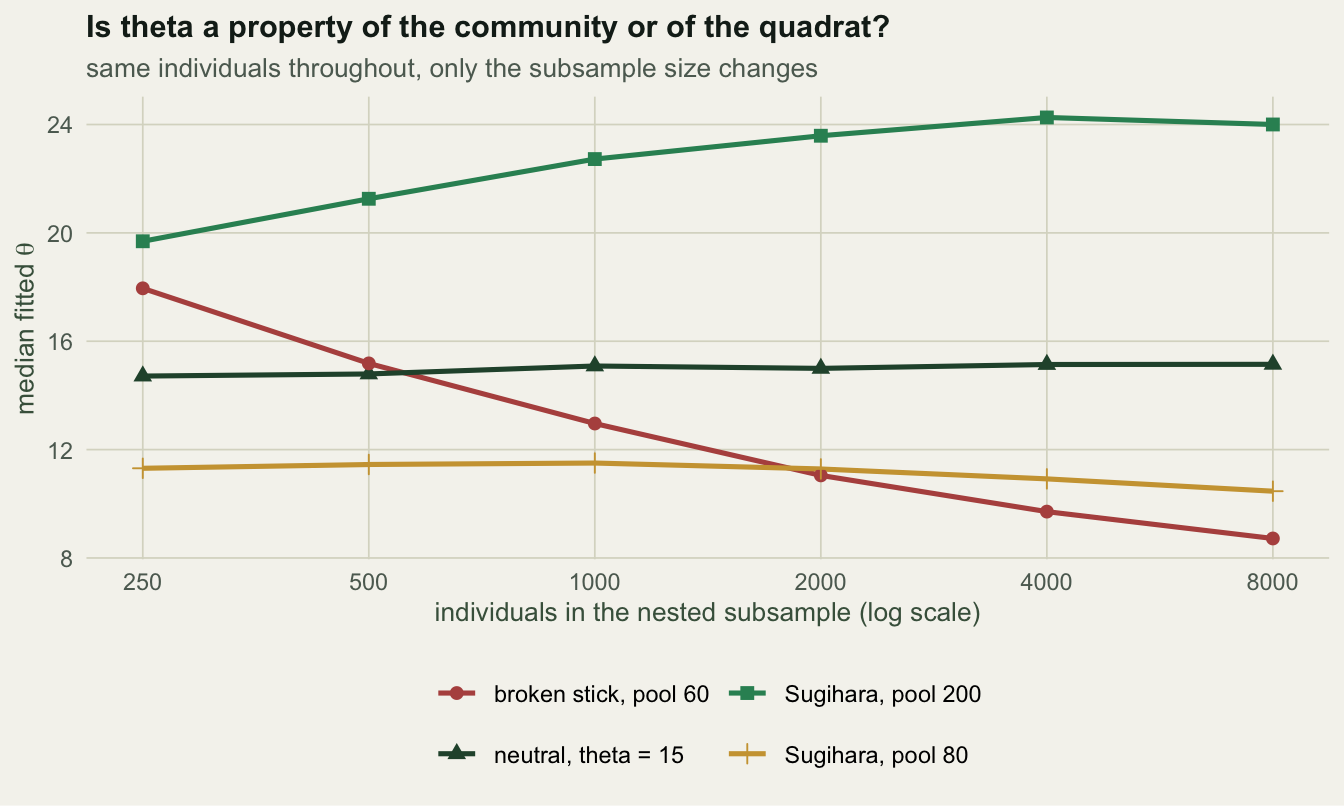
row.names = NULL) model theta_at_250 theta_at_8000 ratio predicted_S observed_S
1 neutral, theta = 15 14.7 15.1 1.00 93 96
2 broken stick, pool 60 18.0 8.7 0.49 110 60
3 Sugihara, pool 80 11.3 10.5 0.92 75 70
4 Sugihara, pool 200 19.7 24.0 1.21 119 140
error within_10pct
1 -2% 55%
2 +83% 0%
3 +7% 38%
4 -15% 32%The neutral row is the calibration: fitted \(\theta\) holds flat across a thirty-two-fold change in sample size, ratio 1.00, and the out-of-sample prediction lands -2% from the observed richness. The check passes where it must.
The broken stick row is the check working. A community with a pool of sixty species cannot keep producing new ones, so its fitted \(\theta\) falls by half as the sample grows, ratio 0.49, and the prediction from the small subsample overshoots by +83%: it expects 110 species where 60 exist. That is the diagnostic doing exactly the job it was built for. The Ewens formula has richness growing like \(\theta \log J\) without limit, and every real species pool is finite, so the mismatch is structural rather than statistical.
Check four: what no amount of data will identify
Some things are not a power problem. From the first post, \(\theta = 2 J_M \nu\) enters the likelihood only as a product, so the metacommunity size and the speciation rate are exactly non-identifiable at any sample size, forever. That one needs no simulation; it is visible in the algebra.
Adding \(m\) creates a softer version of the same problem, and softer means it has to be measured rather than read off. Take one simulated plot, move \(\theta\) away from its estimate, reprofile \(m\) at each new value, and see what it costs.
d <- sort(sim_etienne(th0, I0, Jloc), decreasing = TRUE)
LKd <- logK(d, log_stirling1(max(d)))
f0 <- fit2(d, LKd)
prof_m <- function(th) { # a legjobb m az adott theta mellett
li <- optimize(function(l) -etienne_loglik(d, th, exp(l), LKd), c(log(0.05), log(1e6)))$minimum
exp(li) / (exp(li) + Jloc - 1)
}
same <- data.frame(theta = f0$theta * c(1, 3, 10, 1000, 1 / 3, 1 / 10))
same$m_profiled <- round(sapply(same$theta, prof_m), 4)
same$logL_drop <- round(mapply(function(th, mm) etienne_loglik(d, th, m_to_I(mm, Jloc), LKd),
same$theta, same$m_profiled) - f0$logL, 3)
same$rejected_at_95 <- ifelse(same$logL_drop < -qchisq(0.95, 1) / 2, "yes", "no")
same$theta <- signif(same$theta, 4)
same theta m_profiled logL_drop rejected_at_95
1 26.040 0.0912 0.000 no
2 78.130 0.0251 -1.017 no
3 260.400 0.0167 -2.726 yes
4 26040.000 0.0140 -3.890 yes
5 8.681 0.9990 -8.885 yes
6 2.604 0.9990 -49.891 yesThe asymmetry is the finding. Multiplying \(\theta\) by three costs 1.02 log-likelihood units, well inside the 1.92 that marks the edge of a 95% interval, so the data cannot reject a metacommunity three times as diverse. Dividing it by three costs 8.88, which they can. The likelihood has a wall on the low side and a long shallow slope on the high side, and \(m\) absorbs the slope: across the two rows the data accept, it slides from 0.091 to 0.025 while \(\theta\) triples. The last two rows are pinned against the bound of the search, which is itself the answer: to explain this plot with a \(\theta\) that small, the model needs every individual to be an immigrant.
This is the same object as the 33% of unbounded intervals from check one, seen from the side. Whenever you see a fitted \(\theta\) with no upper interval limit, this is what produced it, and reporting the point estimate alone hides it completely.
Honest limits
These checks test the sampling formula, not the theory. Etienne, Alonso and McKane (2007) showed that Hubbell’s zero-sum constraint can be dropped without altering the sampling formula. So a plot that passes every check here has told you nothing about zero-sum dynamics, because the formula never encoded them.
Snapshots cannot test dynamics. Neutral theory is a claim about birth, death and dispersal rates being equal across species. Nothing above uses a rate. Repeated censuses of the same plot, which give you turnover, are a different and much sharper instrument, and they are where the theory has actually been in trouble.
The blind spot is a warning about check design, not about these two checks. Both diagnostics here reduce a community to a shape. Adding a third one that reads the same shape would add nothing. What would help is a check that reads something else: spatial arrangement, species identity through time, or trait data. Etienne (2007) took the first of those routes.
Where to go next
If the checking pattern is what you came for rather than neutral theory, the same three moves apply to any fitted model: simulate at a known truth and count coverage, predict something the fit was not shown, and enumerate what the likelihood cannot separate. The other checking posts in this blog run the same routine on different machinery.
References
- Etienne 2005 Ecology Letters 8(3):253-260 (10.1111/j.1461-0248.2004.00717.x)
- Etienne 2007 Ecology Letters 10(7):608-618 (10.1111/j.1461-0248.2007.01052.x)
- Etienne, Alonso & McKane 2007 Journal of Theoretical Biology 248(3):522-536 (10.1016/j.jtbi.2007.06.010)
- Kingman 1978 Journal of the London Mathematical Society s2-18(2):374-380 (10.1112/jlms/s2-18.2.374)
- Chave 2004 Ecology Letters 7(3):241-253 (10.1111/j.1461-0248.2003.00566.x)
- McGill et al 2007 Ecology Letters 10(10):995-1015 (10.1111/j.1461-0248.2007.01094.x)
- Rosindell, Hubbell & Etienne 2011 Trends in Ecology and Evolution 26(7):340-348 (10.1016/j.tree.2011.03.024)