m <- 200; m1 <- 40; m0 <- m - m1; mu <- 3; q <- 0.05
nonnull <- c(rep(TRUE, m1), rep(FALSE, m0))
simz <- function(rho){
e <- rnorm(m); g <- rnorm(1)
sqrt(1 - rho) * e + sqrt(rho) * g + ifelse(nonnull, mu, 0)
}Dependence and the Benjamini-Yekutieli correction
R
multiple testing
false discovery rate
ecology tutorial
Benjamini-Hochberg assumes tests are independent or positively dependent. When ecological tests co-vary, Benjamini-Yekutieli is the safe choice, in base R.
The Benjamini-Hochberg procedure controls the false discovery rate, but its proof needs an assumption: the tests must be independent, or positively dependent in a specific sense. Ecological tests rarely are. Taxa co-vary because they share habitat and respond to the same gradients; sites are spatially autocorrelated; abundances are linked through a fixed total. This post asks what happens to BH when the tests are correlated, and introduces the Benjamini-Yekutieli correction, which holds under any dependence at all, in base R.
Setup
What Benjamini-Hochberg assumes
The BH guarantee holds under independence and, more generally, under positive regression dependence (the PRDS condition of Benjamini and Yekutieli 2001). Positively correlated normal test statistics, the natural model for co-varying taxa, satisfy PRDS, so BH still controls the false discovery rate. The catch is what dependence does to the variance of the outcome, and the fact that you cannot verify PRDS from the p-values themselves.
To see it, we build correlated test statistics with a shared latent factor. For an exchangeable correlation rho, setting z_i = sqrt(1 - rho) * e_i + sqrt(rho) * g with independent e_i and a single shared g gives every pair a correlation of exactly rho. (For an arbitrary covariance you would draw from MASS::mvrnorm; the shared-factor trick is the fast special case.)
Positive dependence keeps the mean, spreads the variance
Run the screen three thousand times under independence and under strong positive dependence, and record the realised false discovery proportion each time.
run_bh <- function(rho, seed, R = 3000){
set.seed(seed); fdp <- numeric(R)
for(r in 1:R){
z <- simz(rho); p <- 2 * pnorm(-abs(z))
rb <- BH(p, q)
fdp[r] <- if(sum(rb) == 0) 0 else sum(rb & !nonnull) / sum(rb)
}
fdp
}
fdp_ind <- run_bh(0.0, seed = 808)
fdp_dep <- run_bh(0.5, seed = 809)
c(FDR_independent = round(mean(fdp_ind), 4), SD_independent = round(sd(fdp_ind), 3),
FDR_dependent = round(mean(fdp_dep), 4), SD_dependent = round(sd(fdp_dep), 3))FDR_independent SD_independent FDR_dependent SD_dependent
0.0389 0.0380 0.0351 0.1380 Both mean rates sit under q: BH keeps its false discovery rate promise under positive dependence, exactly as the theory says. But the standard deviation of the realised proportion jumps from 0.038 to 0.138, a 3.6-fold increase. When the shared factor is large, many null taxa cross the threshold together, so false positives arrive in clumps. The rate is controlled on average, but any single study can be much further from q than the independent case would suggest.
d <- data.frame(FDP = c(fdp_ind, fdp_dep),
dep = rep(c("independent","positive dependence (rho=0.5)"), each = length(fdp_ind)))
ggplot(d, aes(FDP, fill = dep, colour = dep)) +
geom_density(alpha = .35, linewidth = .7, adjust = 1.3) +
geom_vline(xintercept = q, linetype = "dashed", colour = te_faint) +
scale_fill_manual(values = c(c_bh, c_raw), name = NULL) +
scale_colour_manual(values = c(c_bh, c_raw), name = NULL) +
coord_cartesian(xlim = c(0, 0.35)) +
labs(x = "realised false discovery proportion (per study)", y = "density",
title = "Dependence keeps BH's mean FDR but inflates its variance") +
theme_te() + theme(legend.position = c(.72, .8))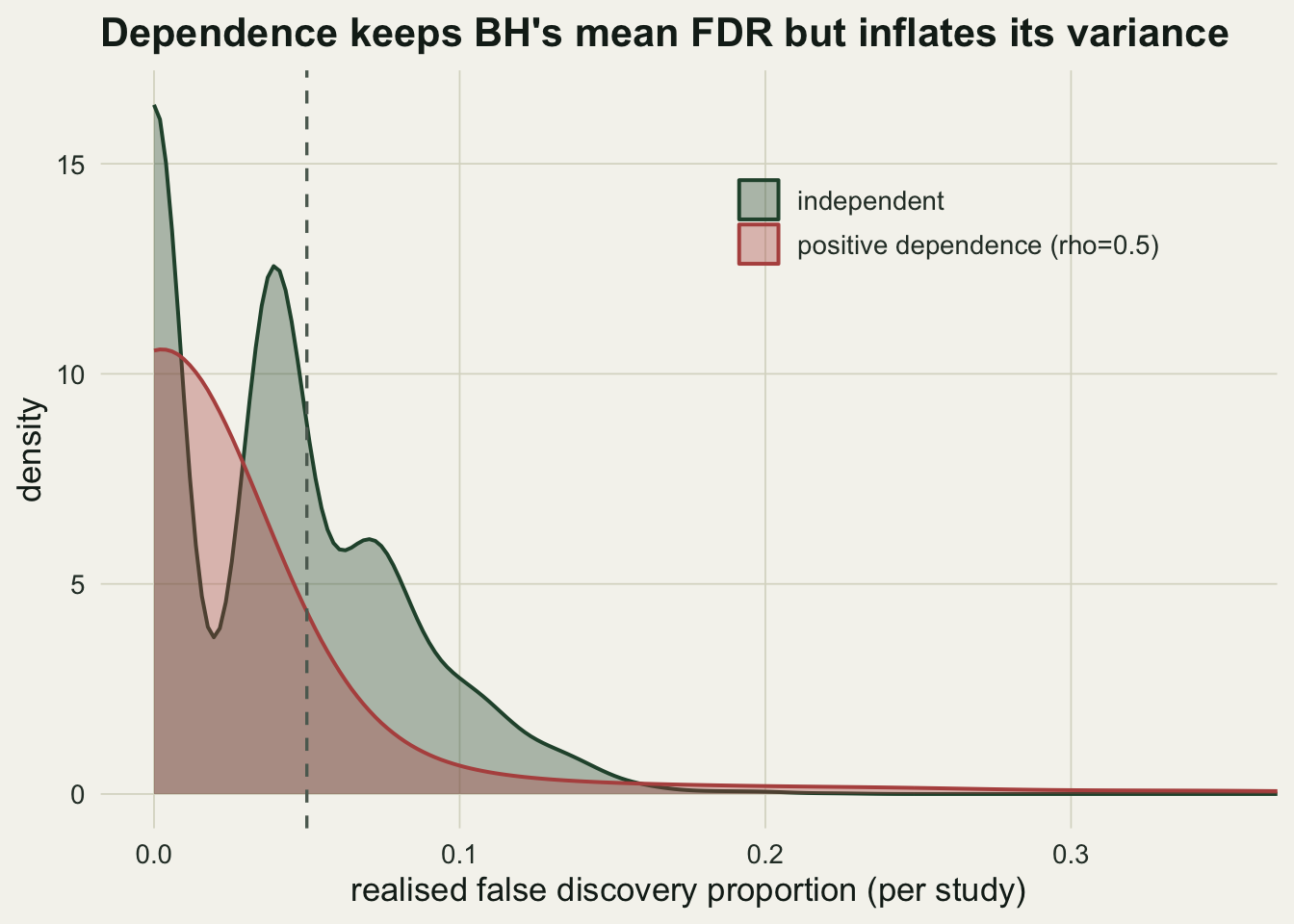
Benjamini-Yekutieli: safe under any dependence
When you cannot justify PRDS, for example under negative dependence or an unknown structure, Benjamini and Yekutieli (2001) give a correction that holds under any dependence. It is BH with the threshold divided by the harmonic number c(m) = 1 + 1/2 + ... + 1/m.
BY <- function(p, q = 0.05){
m <- length(p); cm <- sum(1 / (1:m)); o <- order(p); s <- p[o]
below <- s <= (seq_len(m) / (m * cm)) * q
k <- if(any(below)) max(which(below)) else 0
rej <- rep(FALSE, m); if(k > 0) rej[o[seq_len(k)]] <- TRUE
rej
}
cm <- sum(1 / (1:m))
set.seed(5); pc <- runif(60)^1.5
c(harmonic_cm = round(cm, 3), matches_p.adjust = identical(BY(pc, 0.1), p.adjust(pc, "BY") <= 0.1)) harmonic_cm matches_p.adjust
5.878 1.000 For m = 200 the penalty is c(m) = r round(cm, 3), close to log(m) + 0.577. That makes the Benjamini-Yekutieli threshold almost six times stricter than BH, and the price is power.
run_both <- function(rho, seed, R = 3000){
set.seed(seed); pow_bh <- pow_by <- fdr_by <- numeric(R)
for(r in 1:R){
z <- simz(rho); p <- 2 * pnorm(-abs(z))
rb <- BH(p, q); ry <- BY(p, q)
pow_bh[r] <- mean(rb[nonnull]); pow_by[r] <- mean(ry[nonnull])
fdr_by[r] <- if(sum(ry) == 0) 0 else sum(ry & !nonnull) / sum(ry)
}
c(power_bh = mean(pow_bh), power_by = mean(pow_by), fdr_by = mean(fdr_by))
}
res <- run_both(0.0, seed = 808)
round(res, 4)power_bh power_by fdr_by
0.6053 0.3290 0.0068 Under independence BY holds the false discovery rate at 0.0068, far below the target, and its power is 0.329 against BH’s 0.605: roughly 0.54 of the detections. That is the trade. BY protects you when the dependence is unknown or adversarial, but if your tests really are independent or positively dependent, it leaves a lot of real signal on the table.
db <- data.frame(method = factor(c("BH","BY"), levels = c("BH","BY")),
power = c(res["power_bh"], res["power_by"]),
fdr = c(mean(fdp_ind), res["fdr_by"]))
ggplot(db, aes(method, power, fill = method)) +
geom_col(width = .55) +
geom_text(aes(label = sprintf("power %.2f\nFDR %.3f", power, fdr)),
vjust = -0.2, size = 3.3, colour = te_ink, lineheight = .95) +
scale_fill_manual(values = c(c_bh, c_by), guide = "none") +
scale_y_continuous(limits = c(0, 0.8), expand = expansion(mult = c(0, .18))) +
labs(x = NULL, y = "power",
title = "Benjamini-Yekutieli is safe under any dependence, at a power cost") +
theme_te()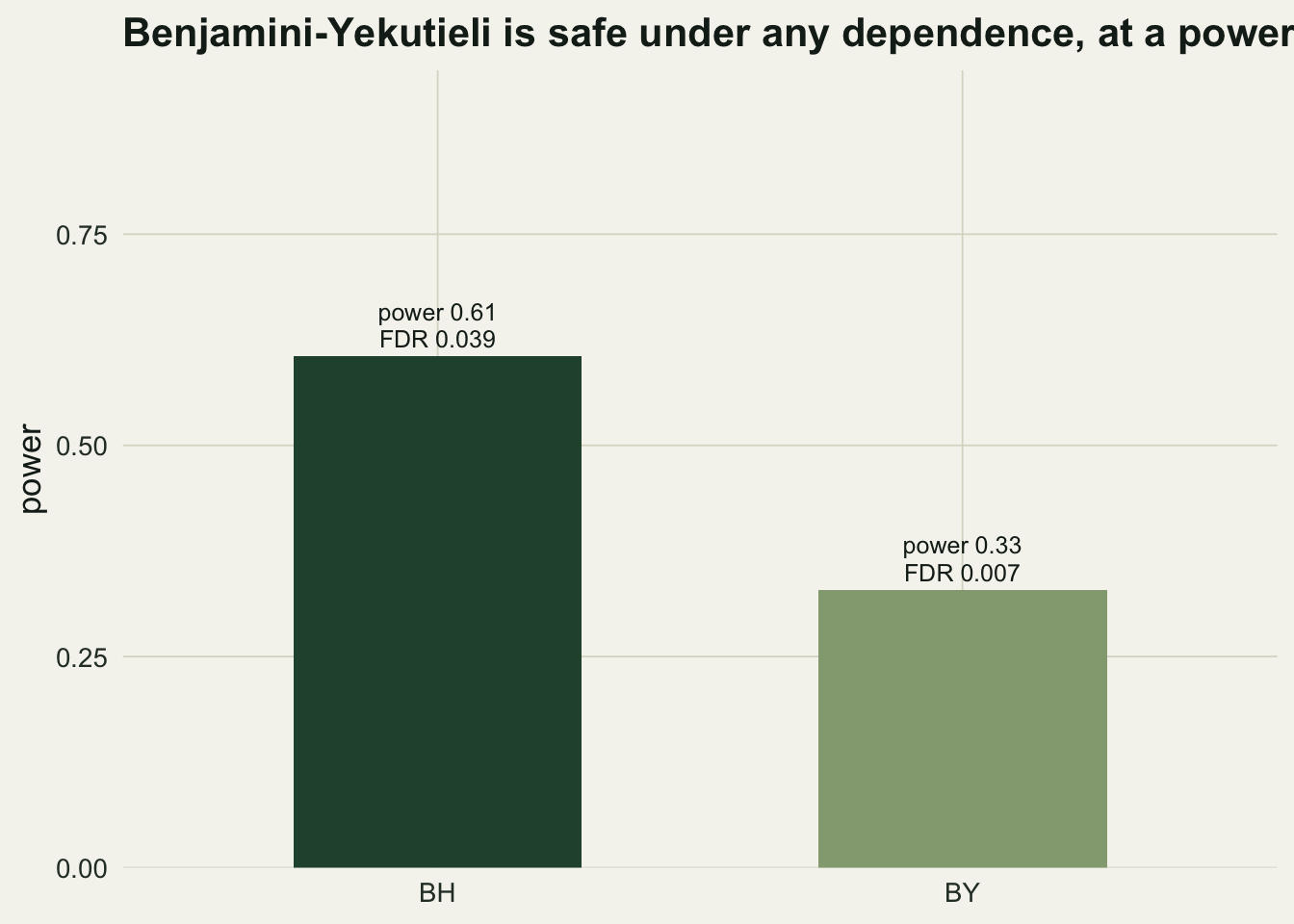
The honest limit
Which correction is right depends on the dependence structure, and that is the part you cannot see. The p-values alone do not reveal whether the tests are independent, positively dependent, or something worse; PRDS is an assumption about the joint distribution of the statistics, argued from how the data were generated, not read off the output. If you can defend positive dependence, BH is efficient and its rate holds. If you cannot, BY is the honest default, bought at a real cost in power. Either way, the dependence between your tests is external information you bring to the analysis, in the same way that a missingness mechanism or a measurement-error variance has to come from outside the data that exhibit it. The final tutorial in this series turns to what you can actually check.
References
Benjamini Y, Yekutieli D 2001. Ann Stat 29(4):1165-1188 (10.1214/aos/1013699998).
Benjamini Y, Hochberg Y 1995. J R Stat Soc Ser B 57(1):289-300 (10.1111/j.2517-6161.1995.tb02031.x).
Benjamini Y, Krieger AM, Yekutieli D 2006. Biometrika 93(3):491-507 (10.1093/biomet/93.3.491).
Reiner A, Yekutieli D, Benjamini Y 2003. Bioinformatics 19(3):368-375 (10.1093/bioinformatics/btf877).
Efron B 2007. J Am Stat Assoc 102(477):93-103 (10.1198/016214506000001211).