alpha <- 0.05
m_grid <- c(1, 5, 10, 20, 50, 100)
fwer <- 1 - (1 - alpha)^m_grid
setNames(round(fwer, 3), m_grid) 1 5 10 20 50 100
0.050 0.226 0.401 0.642 0.923 0.994 Run one test at the five per cent level and you accept a one-in-twenty chance of a false positive. Run twenty tests and that chance is no longer one in twenty. Ecologists reach the many-test situation constantly: a difference tested across every taxon in a table, every pairwise comparison between sites, every candidate driver in a screen. This post shows how fast the error compounds, and works through the corrections that hold it in check, all in base R.
If you run m independent tests and every null is true, the probability that at least one comes back significant at level alpha is one minus the probability that none does:
\[\text{FWER} = 1 - (1-\alpha)^m.\]
This is the family-wise error rate: the chance of one or more false positives across the whole family of tests.
alpha <- 0.05
m_grid <- c(1, 5, 10, 20, 50, 100)
fwer <- 1 - (1 - alpha)^m_grid
setNames(round(fwer, 3), m_grid) 1 5 10 20 50 100
0.050 0.226 0.401 0.642 0.923 0.994 At m = 20 the family-wise error is already 0.642, and by m = 100 it is 0.994. A screen across a hundred taxa is almost guaranteed to hand you a “significant” result even when nothing is going on.
mg <- seq(1, 120)
ggplot(data.frame(m=mg, fwer=1-(1-alpha)^mg), aes(m, fwer)) +
geom_hline(yintercept=alpha, linetype="dashed", colour=te_faint) +
geom_line(colour=te_forest, linewidth=1.1) +
geom_point(data=data.frame(m=20, fwer=1-(1-alpha)^20), size=3, colour=c_raw) +
annotate("text", x=20, y=1-(1-alpha)^20+.06, label="m=20: 0.64",
colour=c_raw, hjust=0, size=3.6) +
scale_y_continuous(limits=c(0,1)) +
labs(x="number of tests (m)", y="family-wise error rate",
title="Running m tests at 0.05 inflates the family-wise error") +
theme_te()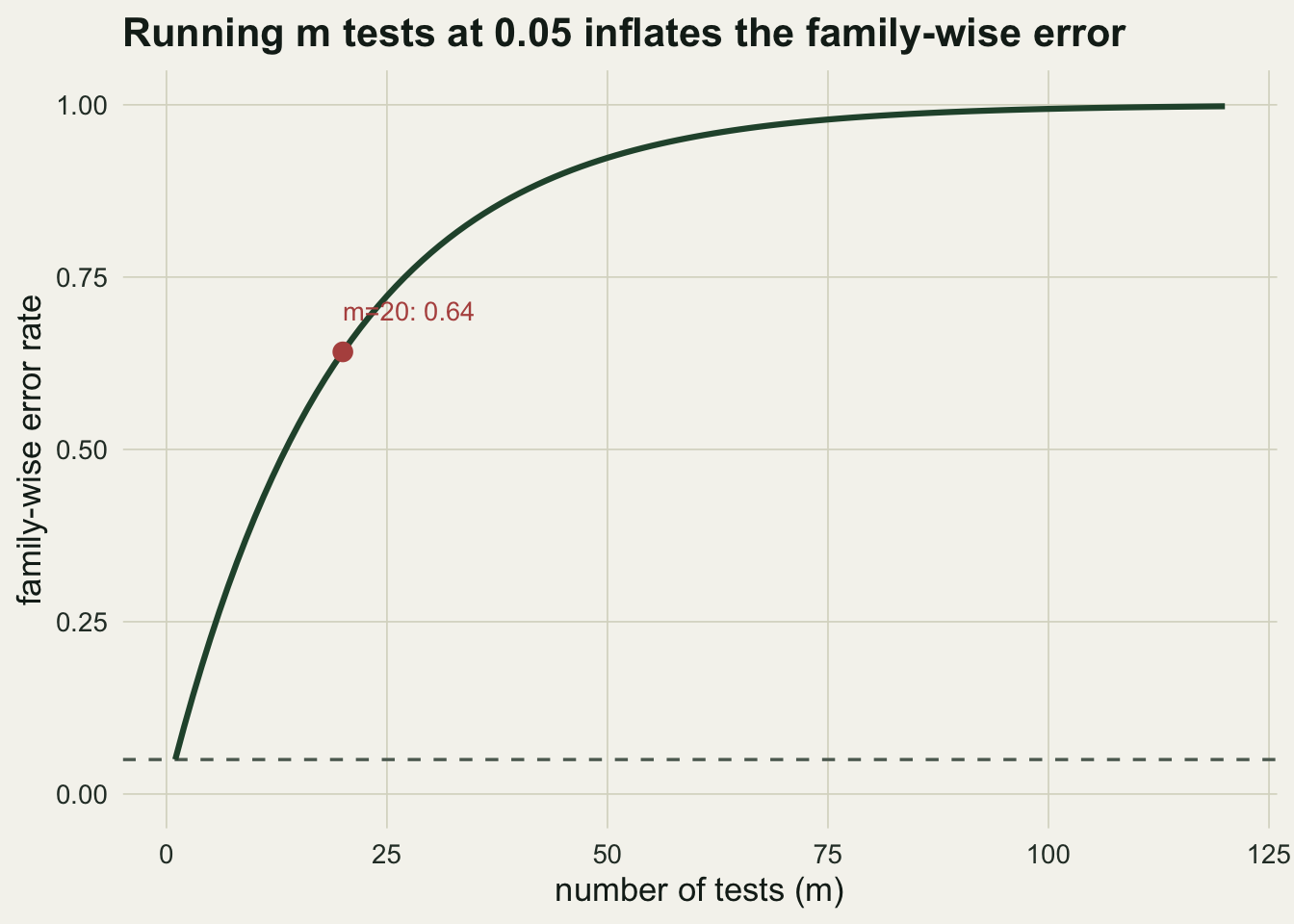
To see the trap in action, simulate twenty taxa measured in two habitats with 30 replicates each, drawn from the same distribution so that every null is genuinely true. We test each taxon with a two-sample t-test.
set.seed(1)
mtax <- 20; n <- 30
grpA <- matrix(rnorm(mtax*n), n, mtax) # habitat A
grpB <- matrix(rnorm(mtax*n), n, mtax) # habitat B, identical distribution
pvals <- sapply(1:mtax, function(j) t.test(grpA[,j], grpB[,j])$p.value)
raw_hits <- which(pvals < alpha)
raw_hits[1] 2 14round(sort(pvals)[1:3], 4)[1] 0.0242 0.0309 0.0820Nothing differs, yet the raw analysis flags 2 taxa (numbers 2 and 14), with the smallest p-value at 0.0242. These are pure false positives. Reporting them as findings is the multiple-comparisons mistake.
The Bonferroni correction tests each hypothesis at alpha / m instead of alpha. It bounds the family-wise error at alpha under any dependence structure, at the price of a very small per-test threshold. Holm’s step-down procedure is a sharper version: sort the p-values, and compare the k-th smallest against alpha / (m - k + 1). It controls the same family-wise error but rejects at least as much as Bonferroni, so there is no reason to prefer plain Bonferroni over it. Sidak’s threshold, 1 - (1 - alpha)^(1/m), is exact under independence.
Each is a few lines by hand, and each matches R’s built-in p.adjust.
bonf <- function(p) pmin(p * length(p), 1)
holm <- function(p){
m <- length(p); o <- order(p)
adj <- cummax((m - seq_len(m) + 1) * p[o])
out <- numeric(m); out[o] <- pmin(adj, 1); out
}
sidak_thr <- 1 - (1 - alpha)^(1/mtax)
c(bonferroni = max(abs(bonf(pvals) - p.adjust(pvals, "bonferroni"))),
holm = max(abs(holm(pvals) - p.adjust(pvals, "holm"))))bonferroni holm
0 0 Applied to the run above, Bonferroni uses a threshold of alpha / m = r alpha/mtax, and the Sidak threshold is 0.00256. Both leave 0 taxa significant, and so does Holm: the two spurious hits are gone.
A short Monte Carlo makes the guarantee concrete. Generate the complete-null scenario three thousand times and record, for each method, whether at least one of the twenty tests is rejected.
welch_p <- function(a, b){
n1<-nrow(a); n2<-nrow(b); m1<-colMeans(a); m2<-colMeans(b)
v1<-apply(a,2,var); v2<-apply(b,2,var); se2<-v1/n1+v2/n2
tt<-(m1-m2)/sqrt(se2)
df<-se2^2/((v1/n1)^2/(n1-1)+(v2/n2)^2/(n2-1))
2*pt(-abs(tt), df)
}
sidak_p <- function(p) pmin(1 - (1-p)^length(p), 1)
set.seed(4242)
R <- 3000
fw <- c(raw=0, bonferroni=0, holm=0, sidak=0)
for(r in 1:R){
A<-matrix(rnorm(mtax*n),n,mtax); B<-matrix(rnorm(mtax*n),n,mtax)
p<-welch_p(A,B)
fw["raw"] <- fw["raw"] + (min(p) < alpha)
fw["bonferroni"] <- fw["bonferroni"] + (min(p.adjust(p,"bonferroni")) < alpha)
fw["holm"] <- fw["holm"] + (min(p.adjust(p,"holm")) < alpha)
fw["sidak"] <- fw["sidak"] + (min(sidak_p(p)) < alpha)
}
round(fw / R, 3) raw bonferroni holm sidak
0.636 0.047 0.047 0.048 The uncorrected analysis fires on 64 per cent of runs, tracking the 0.642 predicted by the formula. Bonferroni, Holm and Sidak all sit at or below the nominal five per cent.
Control is not free. Shrinking the per-test threshold makes real effects harder to detect. Here five of the twenty taxa truly differ (a moderate effect of 0.8 standard deviations), and we track both the power to detect them and the family-wise error among the fifteen nulls.
set.seed(31337)
m1 <- 5; delta <- 0.8
true_eff <- c(rep(TRUE, m1), rep(FALSE, mtax - m1))
mu_vec <- rep(c(delta, 0), c(m1, mtax - m1))
Rp <- 3000
pw <- matrix(0, Rp, 3); fwv <- matrix(0, Rp, 3)
holm_never_worse <- TRUE
for(r in 1:Rp){
A <- matrix(rnorm(mtax*n), n, mtax)
B <- sweep(matrix(rnorm(mtax*n), n, mtax), 2, mu_vec, "+")
p <- welch_p(A, B)
rj <- cbind(p < alpha, p.adjust(p,"bonferroni") < alpha, p.adjust(p,"holm") < alpha)
if(sum(rj[,3]) < sum(rj[,2])) holm_never_worse <- FALSE
pw[r,] <- colMeans(rj[true_eff, , drop=FALSE])
fwv[r,] <- apply(rj[!true_eff, , drop=FALSE], 2, any)
}
power <- setNames(round(colMeans(pw), 3), c("raw","bonferroni","holm"))
fwerp <- setNames(round(colMeans(fwv), 3), c("raw","bonferroni","holm"))
rbind(power = power, fwer = fwerp) raw bonferroni holm
power 0.861 0.486 0.499
fwer 0.533 0.034 0.040d <- data.frame(
method = factor(rep(c("raw (p<0.05)","Bonferroni","Holm"), 2),
levels=c("raw (p<0.05)","Bonferroni","Holm")),
metric = rep(c("power (true effects)","FWER (>=1 false +)"), each=3),
value = c(power, fwerp))
ggplot(d, aes(method, value, fill=method)) +
geom_col(width=.68) + facet_wrap(~metric) +
geom_text(aes(label=sprintf("%.2f", value)), vjust=-0.35, size=3.4, colour=te_ink) +
scale_fill_manual(values=c(c_raw,c_bonf,c_holm), guide="none") +
scale_y_continuous(limits=c(0,1), expand=expansion(mult=c(0,.12))) +
labs(x=NULL, y=NULL, title="Correction trades a little power for controlled error") +
theme_te() + theme(axis.text.x=element_text(angle=12, hjust=1))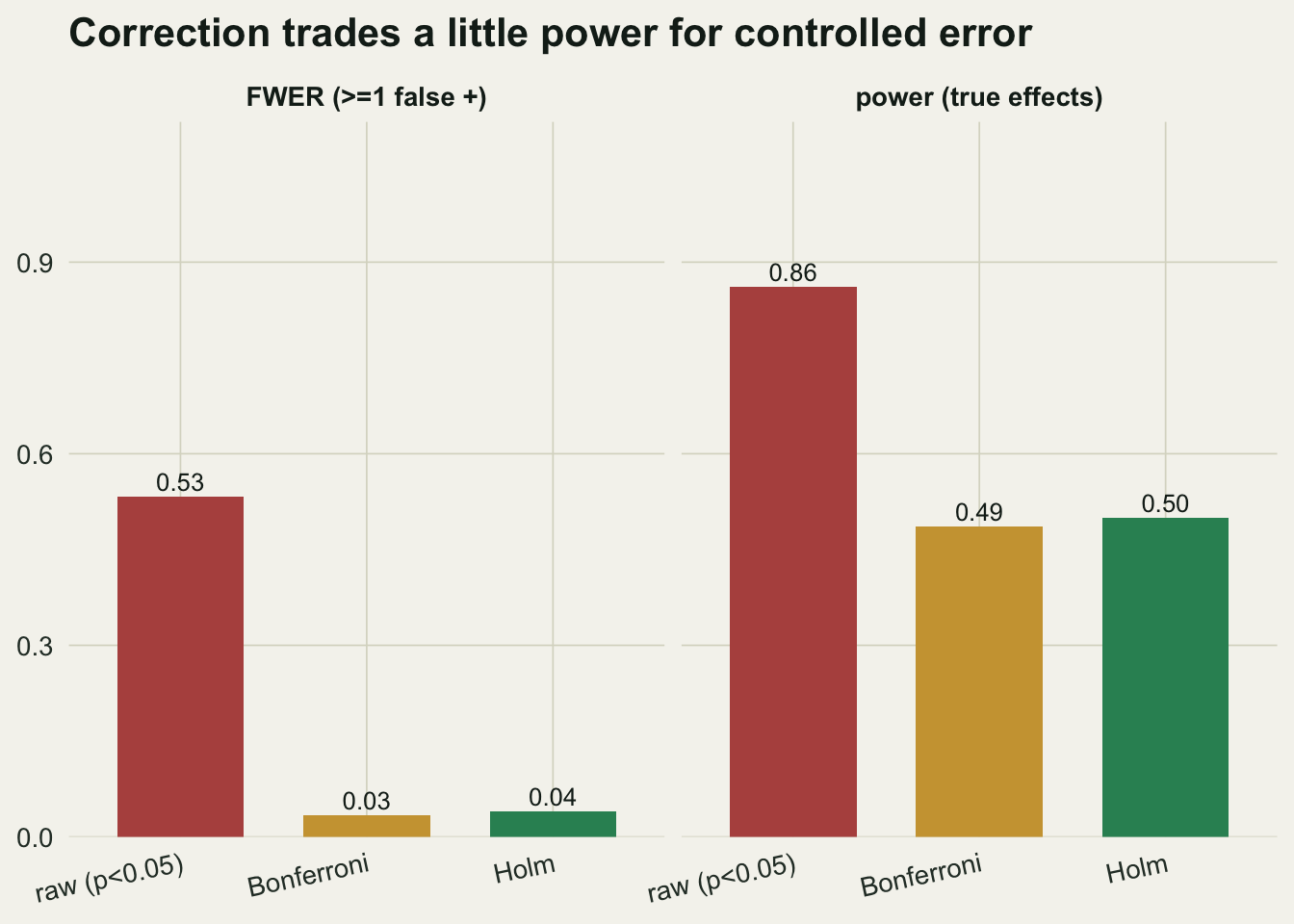
The raw analysis has the highest power (0.861) but a family-wise error of 0.533: in most runs it also flags at least one of the fifteen nulls. Bonferroni cuts power to 0.486 while pinning the error near five per cent. Holm recovers a little of that power (0.499) at the same error, and in this simulation it rejected at least as many hypotheses as Bonferroni in every single run (TRUE), as the theory promises.
The family-wise error rate is a strict target: it guards against even one false positive anywhere in the family. That is the right stance when a single wrong call is costly, but it is often too strict for exploratory ecology, where you scan hundreds of taxa and can tolerate a few false leads if it means finding the real ones. Holm sharpens Bonferroni without changing what it controls; neither tells you which of your significant results are the true ones. The deeper choice is which error rate to control at all. When the family is large and the goal is discovery, the false discovery rate is usually the better currency, and that is the subject of the next tutorial.
Benjamini Y, Hochberg Y 1995. J R Stat Soc Ser B 57(1):289-300 (10.1111/j.2517-6161.1995.tb02031.x).
Holm S 1979. Scand J Stat 6(2):65-70 (no DOI).
Sidak Z 1967. J Am Stat Assoc 62(318):626-633 (10.1080/01621459.1967.10482935).
Aickin M, Gensler H 1996. Am J Public Health 86(5):726-728 (10.2105/AJPH.86.5.726).
Rice WR 1989. Evolution 43(1):223-225 (10.1111/j.1558-5646.1989.tb04220.x).
Moran MD 2003. Oikos 100(2):403-405 (10.1034/j.1600-0706.2003.12010.x).
Nakagawa S 2004. Behav Ecol 15(6):1044-1045 (10.1093/beheco/arh107).
Quinn GP, Keough MJ 2002. Experimental Design and Data Analysis for Biologists. Cambridge University Press. ISBN 978-0-521-00976-8.