library(ggplot2); library(dplyr); library(tidyr)
te <- c(forest = "#2f5d50", moss = "#7a9b76", rust = "#b5651d", gold = "#c9a227",
slate = "#3d4b53", sand = "#d9cbb2", sky = "#5b8aa6", brick = "#8c3b2e")
theme_te <- function(base_size = 12) {
theme_minimal(base_size = base_size) +
theme(panel.grid.minor = element_blank(),
panel.grid.major = element_line(colour = "#e7e1d5", linewidth = 0.3),
axis.title = element_text(colour = "#3d4b53"),
axis.text = element_text(colour = "#5c6670"),
plot.title = element_text(colour = "#2f3a40", face = "bold", size = base_size + 1),
plot.subtitle = element_text(colour = "#5c6670", size = base_size - 1),
legend.position = "bottom",
legend.title = element_text(colour = "#3d4b53"),
plot.background = element_rect(fill = "white", colour = NA),
panel.background = element_rect(fill = "white", colour = NA))
}Coverage-based rarefaction and extrapolation
R
species richness
biodiversity
ecology tutorial
Comparing richness at equal sample size can rank two assemblages the wrong way. Standardise by sample coverage instead, in base R, and see where extrapolation stops.
Two samples, different numbers of individuals, and you want to know which assemblage is richer. The reflex is to rarefy both down to the same number of individuals and compare. That is the right instinct about sample size, but it can still give the wrong answer, because two samples of the same size can differ in how completely they cover their communities. The fix, from Chao and Jost, is to standardise by sample coverage rather than by count. This post builds rarefaction, extrapolation and coverage from scratch and shows a comparison that reverses when you switch bases.
The building blocks
Three functions carry the whole post. Rarefied richness is Hurlbert’s expectation: the mean number of species in a random subsample of m individuals. Rarefied coverage is the expected sample coverage of that subsample. Sample coverage itself, the fraction of the community’s individuals that belong to species you sampled, comes from a Good-Turing formula, improved by Chao and Jost with the doubletons term.
lchoose_safe <- function(n, k) ifelse(k > n | k < 0, -Inf, lchoose(n, k))
rare_S <- function(x, m) # Hurlbert rarefied richness
sapply(m, function(mm) { n <- sum(x)
sum(1 - exp(lchoose_safe(n - x, mm) - lchoose_safe(n, mm))) })
rare_C <- function(x, m) # rarefied sample coverage
sapply(m, function(mm) { n <- sum(x)
1 - sum((x / n) * exp(lchoose_safe(n - x, mm) - lchoose_safe(n - 1, mm))) })
covhat <- function(x) { n <- sum(x); f1 <- sum(x == 1); f2 <- sum(x == 2)
if (f2 == 0) f2 <- f1 * (f1 - 1) / 2
1 - (f1 / n) * ((n - 1) * f1 / ((n - 1) * f1 + 2 * f2)) }
chao1 <- function(x) { n <- sum(x); f1 <- sum(x == 1); f2 <- sum(x == 2)
length(x) + (n - 1) / n * f1 * (f1 - 1) / (2 * (f2 + 1)) }
extr_S <- function(x, m) { n <- sum(x); f1 <- sum(x == 1); f2 <- sum(x == 2)
f0 <- (n - 1) / n * f1 * (f1 - 1) / (2 * (f2 + 1)); So <- length(x)
sapply(m, function(mm) if (mm <= n)
sum(1 - exp(lchoose_safe(n - x, mm) - lchoose_safe(n, mm)))
else So + f0 * (1 - (1 - f1 / (n * f0 + f1))^(mm - n))) }Two assemblages, same effort
Assemblage A is species-poor but even; assemblage B is richer but dominated by a few common species with a long tail of rarities. Both are sampled with 250 individuals.
set.seed(13)
SA <- 90; relA <- exp(rnorm(SA, 0, 0.55)); relA <- relA / sum(relA)
SB <- 200; relB <- exp(rnorm(SB, 0, 1.70)); relB <- relB / sum(relB)
n <- 250
xA <- as.vector(rmultinom(1, n, relA)); xA <- xA[xA > 0]
xB <- as.vector(rmultinom(1, n, relB)); xB <- xB[xB > 0]
SobsA <- length(xA); SobsB <- length(xB); CA <- covhat(xA); CB <- covhat(xB)
round(c(SobsA = SobsA, SobsB = SobsB, coverageA = CA, coverageB = CB), 4) SobsA SobsB coverageA coverageB
74.0000 68.0000 0.9325 0.8524 At equal size the naive comparison is clear. Assemblage A shows 74 species, assemblage B shows 68, so A looks the richer of the two. It is not: A has 90 species in truth and B has 200. The clue is coverage. A is sampled to 0.933 coverage, B only to 0.852: B’s curve is still climbing steeply, its rare tail barely touched, so its 68 is a far worse snapshot of the truth than A’s 74.
Extrapolation shows the curves crossing
Rarefaction runs the sample down; extrapolation runs it up, using the Chao1 estimate of unseen species to project where the curve is heading. Reliable extrapolation reaches only about twice the reference sample size, so we stop at 500.
SA_2 <- extr_S(xA, 2 * n); SB_2 <- extr_S(xB, 2 * n)
c(A_at_500 = round(SA_2, 1), B_at_500 = round(SB_2, 1),
Chao1_A = round(chao1(xA), 1), Chao1_B = round(chao1(xB), 1))A_at_500 B_at_500 Chao1_A Chao1_B
80.7 94.3 81.5 119.0 Projected to 500 individuals, A reaches 80.7 and B reaches 94.3: B has overtaken A. The equal-size verdict was an artefact of stopping at 250 individuals, on the wrong side of where the two curves cross. The Chao1 asymptotes tell the same story, 81.5 for A against 119 for B, though both underestimate the true richness because the rarest species leave no trace in a sample this size.
grid <- seq(1, 2 * n, by = 5)
mk <- function(x, lab) data.frame(m = grid, S = extr_S(x, grid), assemblage = lab,
part = ifelse(grid <= n, "reference", "extrapolation"))
allc <- rbind(mk(xA, "A (even, true S = 90)"), mk(xB, "B (uneven, true S = 200)"))
cols <- c("A (even, true S = 90)" = unname(te["forest"]),
"B (uneven, true S = 200)" = unname(te["rust"]))
ggplot(allc, aes(m, S, colour = assemblage)) +
geom_vline(xintercept = n, linetype = "dotted", colour = te["slate"], linewidth = 0.4) +
geom_line(aes(linetype = part), linewidth = 0.9) +
geom_point(data = data.frame(m = n, S = c(SobsA, SobsB),
assemblage = names(cols)), size = 3, show.legend = FALSE) +
annotate("text", x = n, y = 20, label = "reference sample n = 250",
angle = 90, vjust = -0.4, hjust = 0, size = 3, colour = te["slate"]) +
scale_colour_manual(values = cols, name = NULL) +
scale_linetype_manual(values = c("reference" = "solid", "extrapolation" = "22"), name = NULL) +
labs(title = "At equal sample size A looks richer; the curves cross",
subtitle = "Solid = interpolation, dashed = extrapolation; B overtakes A before twice the effort",
x = "number of individuals", y = "expected species richness") +
theme_te()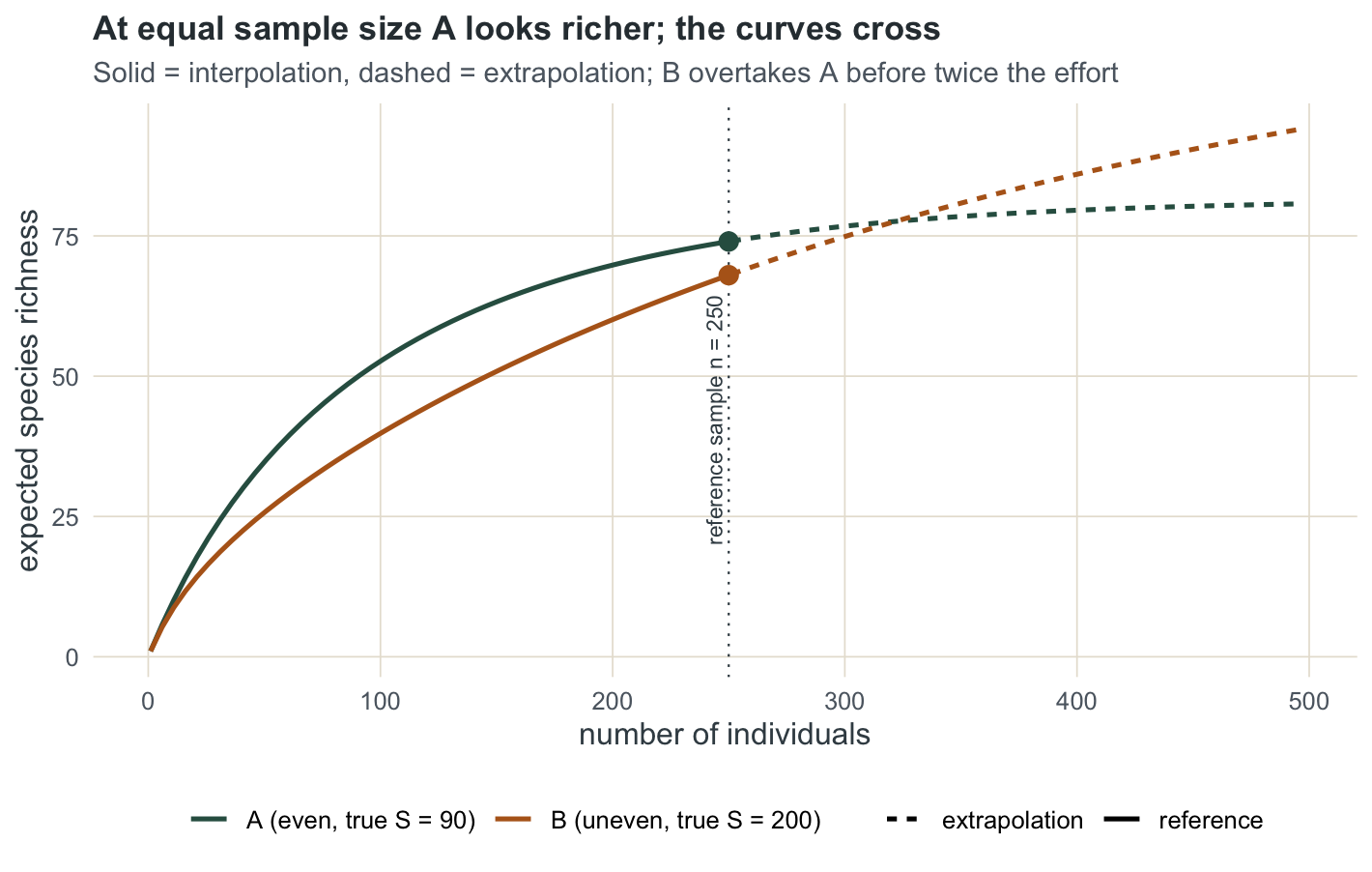
Standardising by coverage reverses the verdict
The principled comparison holds coverage constant, not size. Bring both samples to the coverage of the less complete one, 0.8524, by rarefying A down until it reaches that coverage.
Clow <- min(CA, CB)
m_for_C <- function(x, Ct) { ms <- 1:sum(x); ms[which.min(abs(rare_C(x, ms) - Ct))] }
mA_low <- m_for_C(xA, Clow)
SA_low <- rare_S(xA, mA_low) # A rarefied to B's coverage
SB_low <- SobsB # B is already at this coverage
c(target_coverage = round(Clow, 4), A_richness = round(SA_low, 1),
A_at_m = mA_low, B_richness = round(SB_low, 1))target_coverage A_richness A_at_m B_richness
0.8524 65.0000 161.0000 68.0000 Rarefying A to coverage 0.8524 takes it down to 161 individuals and 65 species, below B’s 68. On equal-coverage footing the ranking flips to B richer than A, agreeing with the extrapolation and with the truth. The equal-size comparison penalised nothing about B being under-sampled; the equal-coverage comparison removes that confound.
ms <- 1:n
cc <- rbind(
data.frame(C = rare_C(xA, ms), S = rare_S(xA, ms), assemblage = "A (even, true S = 90)"),
data.frame(C = rare_C(xB, ms), S = rare_S(xB, ms), assemblage = "B (uneven, true S = 200)"))
ggplot(cc, aes(C, S, colour = assemblage)) +
geom_vline(xintercept = Clow, linetype = "dotted", colour = te["slate"], linewidth = 0.4) +
geom_line(linewidth = 0.9) +
geom_point(data = data.frame(C = c(CA, CB), S = c(SobsA, SobsB),
assemblage = names(cols)), size = 3, show.legend = FALSE) +
annotate("text", x = Clow, y = 15, label = "equal coverage 0.852",
angle = 90, vjust = 1.2, hjust = 0, size = 3, colour = te["slate"]) +
scale_colour_manual(values = cols, name = NULL) +
scale_x_continuous(limits = c(0.3, 0.95)) +
labs(title = "On the coverage axis the ranking reverses",
subtitle = "At equal coverage A (rarefied) falls below B: the fair comparison matches the asymptote",
x = "sample coverage", y = "expected species richness") +
theme_te()Warning: Removed 30 rows containing missing values or values outside the scale range
(`geom_line()`).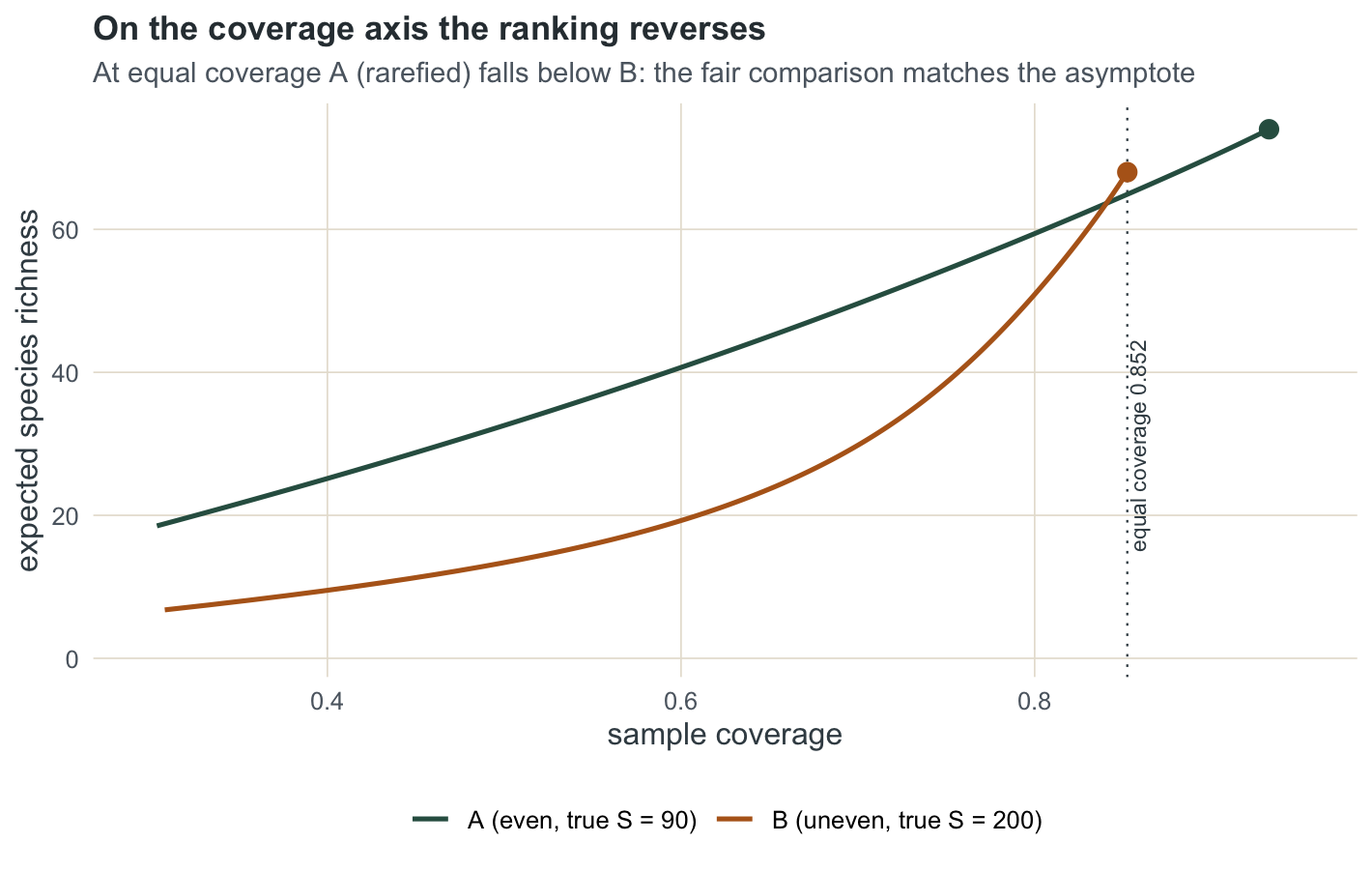
Where it stops
Coverage-based standardisation makes the comparison fair, but it does not conjure the truth. It compares richness up to the shared coverage; it does not recover B’s full 200 species, because those rarest species are simply not in the sample. Extrapolation can reach a little past the data, to roughly twice the sample size, and no further with any confidence: push it and you are reading the estimator’s assumptions, not the community. Comparing samples fairly and estimating true richness are two different jobs, and the second is always bounded by what you sampled. The next tutorial takes the same limit into the wider diversity profile.
References
Hurlbert S H 1971 Ecology 52(4):577-586 (10.2307/1934145)
Good I J 1953 Biometrika 40(3-4):237-264 (10.1093/biomet/40.3-4.237)
Chao A, Jost L 2012 Ecology 93(12):2533-2547 (10.1890/11-1952.1)
Colwell R K, Chao A, Gotelli N J, Lin S-Y, Mao C X, Chazdon R L, Longino J T 2012 Journal of Plant Ecology 5(1):3-21 (10.1093/jpe/rtr044)
Chao A, Gotelli N J, Hsieh T C, Sander E L, Ma K H, Colwell R K, Ellison A M 2014 Ecological Monographs 84(1):45-67 (10.1890/13-0133.1)
Hsieh T C, Ma K H, Chao A 2016 Methods in Ecology and Evolution 7(12):1451-1456 (10.1111/2041-210X.12613)