library(ape)
library(ggplot2)
set.seed(292)
S <- 200
pool_tree <- rcoal(S, tip.label = sprintf("sp%03d", 1:S))
pool_tree$edge.length <- pool_tree$edge.length / max(node.depth.edgelength(pool_tree))
pool <- pool_tree$tip.label
D <- cophenetic(pool_tree)[pool, pool]
trait <- rTraitCont(pool_tree, model = "BM", sigma = 1)[pool]
trait <- (trait - mean(trait)) / sd(trait)Phylogenetic community structure: NRI and NTI in R
R
community phylogenetics
biodiversity
ggplot2
ecology tutorial
Raw MPD and MNTD are confounded with species richness. Build standardised effect sizes, NRI and NTI, by hand in R and learn to read their signs.
You have a species list for a plot and a phylogeny for the regional flora. Are the species in that plot closer relatives than you would expect, and if so, does that say anything about how the community was assembled?
The metric part of this question is easy. The interpretive part is where the whole field lives, and it starts with a fact that is easy to miss: the raw number you compute from a plot is not a property of the plot. It is mostly a property of how many species the plot has.
This post builds the standard fix from scratch: the standardised effect size, and its two sign-flipped aliases, the Net Relatedness Index (NRI) and the Nearest Taxon Index (NTI). Everything is base R plus ape for the tree.
Setup
A regional pool of 200 species on a coalescent tree, scaled so the root sits at depth 1 (the largest possible distance between two tips is therefore 2). One trait evolves on the tree under Brownian motion, so close relatives resemble each other: that is the assumption the whole method rests on, and it is worth naming out loud.
cophenetic() gives the patristic distance matrix: the summed branch length on the path between each pair of tips. Everything below reads from that matrix and nothing else.
Two distances, two depths
Two summaries dominate the literature, and they are two lines each.
mpd <- function(sp) { d <- D[sp, sp]; mean(d[lower.tri(d)]) }
mntd <- function(sp) {
d <- D[sp, sp]
diag(d) <- NA
mean(apply(d, 1, min, na.rm = TRUE))
}Mean pairwise distance (MPD) averages over every pair in the community. Mean nearest taxon distance (MNTD) averages, over each species, the distance to its single closest relative in the community.
They are not two flavours of the same thing. MPD is dominated by the deep splits: swap one species for a member of a distant clade and every pair involving it changes. MNTD only looks at each species’ nearest neighbour, so it is a measurement of the terminal structure. A community can be built from two distant clades (high MPD) with tight species clusters inside each (low MNTD). Keep that picture; it explains most of the confusing results later.
The raw number is mostly a richness measurement
Here is the experiment nobody runs before reporting an MNTD. Draw communities at random from the pool at five richness levels, with no assembly rule whatsoever, and look at what the raw metrics do.
neutral <- function(k) sample(pool, k)
set.seed(1)
kg <- rep(c(4, 8, 15, 25, 40), each = 200)
raw <- t(sapply(kg, function(k) { s <- neutral(k); c(mpd(s), mntd(s)) }))
tab <- data.frame(
k = sort(unique(kg)),
mpd_m = tapply(raw[, 1], kg, mean), mpd_s = tapply(raw[, 1], kg, sd),
mntd_m = tapply(raw[, 2], kg, mean), mntd_s = tapply(raw[, 2], kg, sd)
)
round(tab, 3) k mpd_m mpd_s mntd_m mntd_s
4 4 1.129 0.287 0.634 0.243
8 8 1.129 0.183 0.342 0.135
15 15 1.144 0.091 0.204 0.074
25 25 1.142 0.064 0.129 0.044
40 40 1.150 0.046 0.084 0.026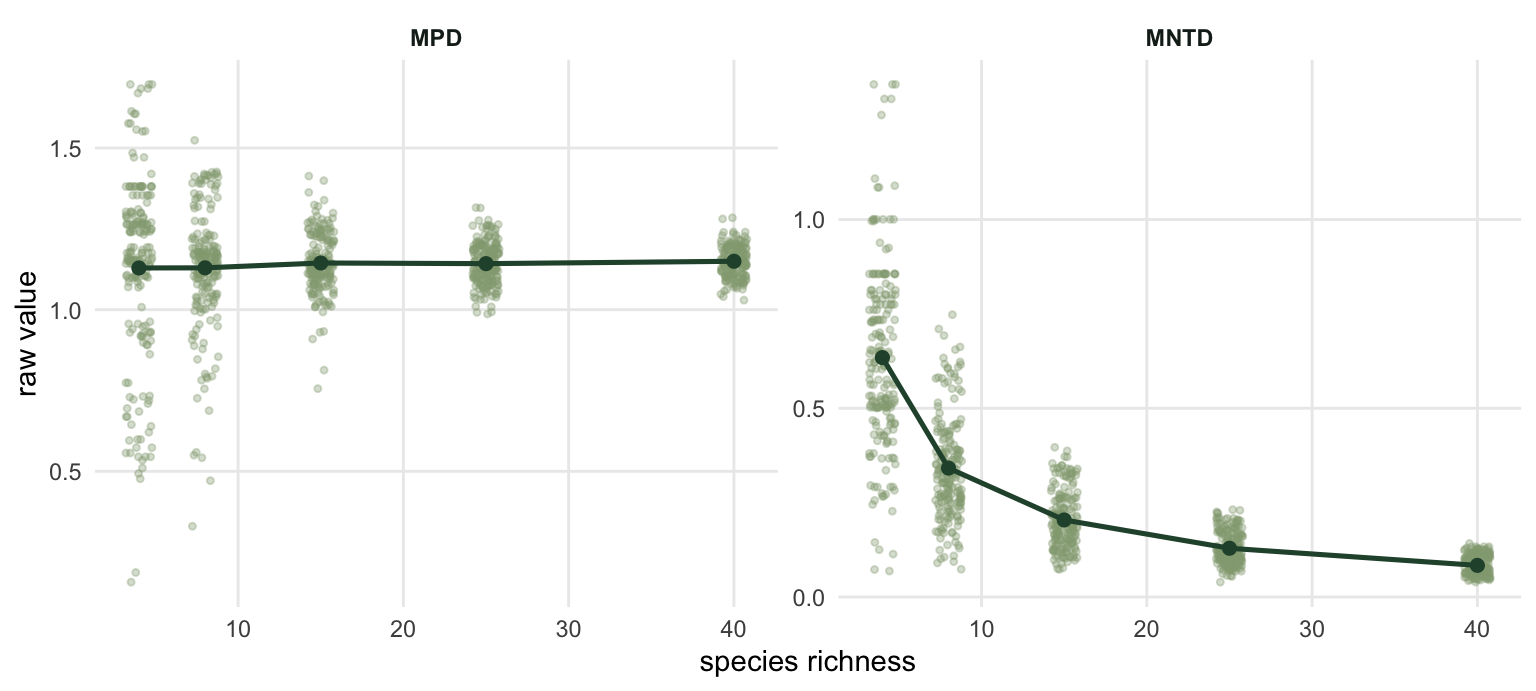
The MNTD mean falls by a factor of 7.57 between richness 4 and richness 40. Nothing ecological happened. Add species to a fixed pool and each species’ nearest relative simply gets nearer, because there are more candidates. Any comparison of raw MNTD between a species-poor plot and a species-rich plot is a comparison of richness wearing a phylogenetic costume.
MPD looks safer at first: the mean barely moves (1.13 to 1.15). But look at the second column. The standard deviation shrinks by a factor of 6.21 over the same range. A four-species plot sitting 0.2 below the pool mean is unremarkable; a forty-species plot sitting 0.2 below it is extraordinary. The raw deviation means different things at different richness, which is the same problem in a quieter voice.
Standardising against a null
The fix is the obvious one. Ask what the metric would look like if this community, with exactly this richness, had been drawn at random from the pool, and express the observation in units of that distribution.
ses <- function(sp, R = 999) {
k <- length(sp)
obs <- c(mpd(sp), mntd(sp))
nul <- replicate(R, { s <- sample(pool, k); c(mpd(s), mntd(s)) })
z <- (obs - rowMeans(nul)) / apply(nul, 1, sd)
p <- (rowSums(nul <= obs) + 1) / (R + 1)
list(obs = obs, null = nul, z = z, p = p, nri = -z[1], nti = -z[2])
}Three conventions are buried in those five lines, and each one has cost somebody a paragraph in review.
The sign is flipped. NRI is defined as minus the standardised MPD, and NTI as minus the standardised MNTD. So positive NRI means clustering: the community is more closely related than chance, which is a low MPD. If you report ses.mpd from picante and then discuss it as if it were NRI, every sign in your discussion is backwards. This is the single most common slip in the method.
The p-value is a rank, not a z-test. The p line counts how many null draws fall at or below the observation. That works whatever shape the null distribution has, which matters more than it sounds; see the checking post for a case where the two disagree.
The null is a decision, not a default. Here the null draws species uniformly from the pool. That is one hypothesis out of several, and it is the subject of the next post.
Three assembly rules, three signatures
Now build communities under three rules and see what the index reports. Habitat filtering picks species whose trait is near a local optimum. Limiting similarity picks species that are far from whatever has already been picked. The neutral rule picks at random.
filtered <- function(k, opt, w = 0.3) {
sample(pool, k, prob = exp(-(trait - opt)^2 / (2 * w^2)))
}
spaced <- function(k) {
sel <- sample(pool, 1)
while (length(sel) < k) {
cand <- setdiff(pool, sel)
sel <- c(sel, sample(cand, 1, prob = apply(D[cand, sel, drop = FALSE], 1, min)^8))
}
sel
}
set.seed(10); cf <- filtered(15, 1.2)
set.seed(11); cn <- neutral(15)
set.seed(12); co <- spaced(15)
set.seed(20); sf <- ses(cf)
set.seed(21); sn <- ses(cn)
set.seed(22); so <- ses(co)
data.frame(
rule = c("filtered", "neutral", "spaced"),
MPD = round(c(sf$obs[1], sn$obs[1], so$obs[1]), 3),
NRI = round(c(sf$nri, sn$nri, so$nri), 2),
MNTD = round(c(sf$obs[2], sn$obs[2], so$obs[2]), 3),
NTI = round(c(sf$nti, sn$nti, so$nti), 2)
) rule MPD NRI MNTD NTI
1 filtered 0.724 4.67 0.029 2.29
2 neutral 1.123 0.20 0.140 0.85
3 spaced 1.294 -1.62 0.433 -3.25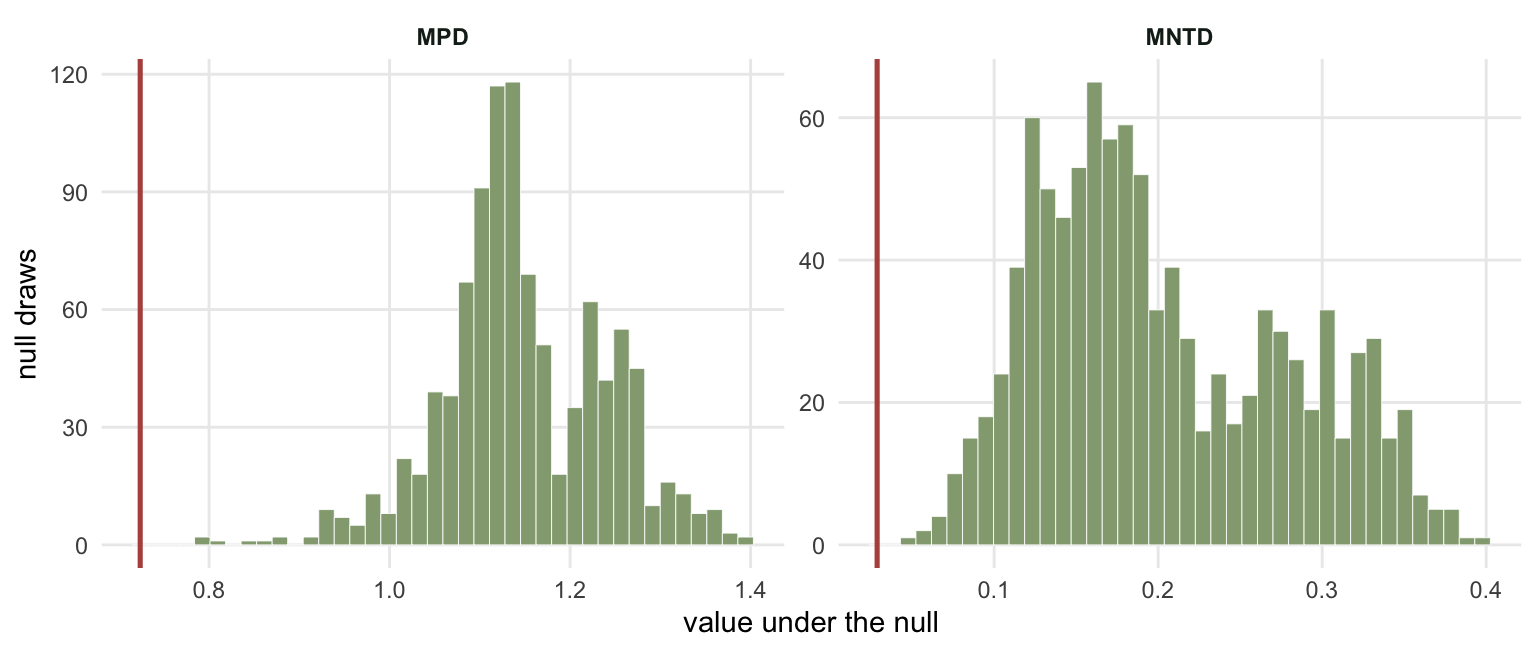
The filtered community lands at NRI +4.67 with a rank p of 0.001: strongly clustered. The neutral community lands at NRI +0.20, p 0.424, which is the correct answer. The spaced community lands at NTI -3.25: overdispersed at the tips.
Repeat over 100 communities per rule to see how reliable that is.
rep_ses <- function(gen, n = 100, R = 199) {
t(replicate(n, {
sp <- gen(15)
obs <- c(mpd(sp), mntd(sp))
nul <- replicate(R, { s <- sample(pool, 15); c(mpd(s), mntd(s)) })
-(obs - rowMeans(nul)) / apply(nul, 1, sd)
}))
}
set.seed(30); A <- rep_ses(function(k) filtered(k, rnorm(1)))
set.seed(31); B <- rep_ses(neutral)
set.seed(32); C <- rep_ses(spaced)
data.frame(
rule = c("filtered", "neutral", "spaced"),
NRI = round(c(mean(A[, 1]), mean(B[, 1]), mean(C[, 1])), 2),
NRI_sd = round(c(sd(A[, 1]), sd(B[, 1]), sd(C[, 1])), 2),
NTI = round(c(mean(A[, 2]), mean(B[, 2]), mean(C[, 2])), 2),
NTI_sd = round(c(sd(A[, 2]), sd(B[, 2]), sd(C[, 2])), 2)
) rule NRI NRI_sd NTI NTI_sd
1 filtered 3.39 3.76 1.33 0.90
2 neutral 0.06 1.09 0.17 1.04
3 spaced -1.57 0.13 -3.09 0.18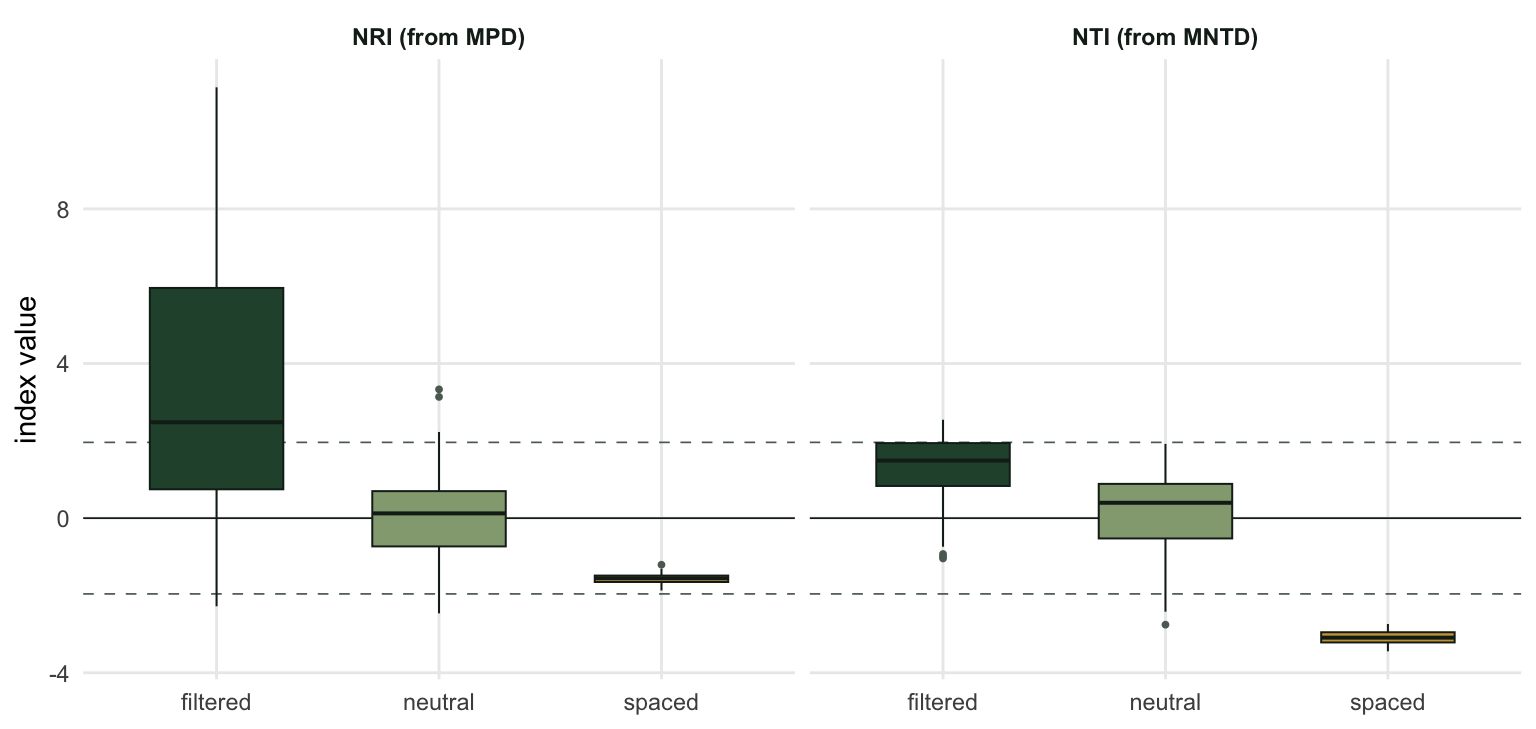
The neutral row is the one to read first. Its NRI averages +0.06 with a spread of 1.09, and its NTI averages +0.17 with a spread of 1.04. Centred on zero, spread of about one: that is what a standardised effect size is supposed to do when the data really were generated by the null. The metric is calibrated.
Why NRI and NTI disagree
Now the asymmetry. Habitat filtering shows up mainly in NRI (+3.39, against +1.33 for NTI). Limiting similarity shows up mainly in NTI (-3.09, against -1.57 for NRI).
That is not a quirk of these simulations. It follows from what the two metrics measure. A conserved trait varies most between distant clades, so filtering on it selects whole clades and collapses the deep structure, which is exactly what MPD sees. A rule that rejects each candidate for being too close to a resident acts on nearest neighbours, which is exactly what MNTD sees. The two indices are not redundant, and a paper reporting only one of them has answered only one of the two questions.
Two more things in that table deserve a mention. The filtered NRI has a spread of 3.76, far wider than the other rules, because a randomly placed trait optimum sometimes lands in a crowded part of the pool and sometimes in an empty one. And the spaced rule produces a very tight distribution, because the greedy algorithm that builds it is nearly deterministic. Effect size and its variability are separate facts about a process.
The honest limit
The number that came out of ses() is not “how clustered this community is”. It is “how clustered this community is relative to a uniform draw from these 200 species”. Change the 200 and the answer changes, and there is no data-internal test that tells you which 200 is right. Everything downstream inherits this. The tree inherits it too: swap the tree and the distances move.
The method is worth using anyway, because the alternative is reading raw MNTDs across plots of unequal richness, which measures nothing at all. But the standardised number is a statement about a comparison you chose, not a discovery about nature. The checking post makes that concrete by moving the pool and watching the conclusion move with it.
Where to go next
The obvious next question is the one this post kept deferring: which null? Drawing uniformly from the pool is not the only option, and the alternatives ask genuinely different questions rather than being better or worse approximations of one question. That comes next. After that, the same tree and the same distance matrix answer a between-community question instead of a within-community one.
References
- Webb 2000 American Naturalist 156(2):145-155 (10.1086/303378)
- Webb, Ackerly, McPeek, Donoghue 2002 Annual Review of Ecology and Systematics 33:475-505 (10.1146/annurev.ecolsys.33.010802.150448)
- Webb, Ackerly, Kembel 2008 Bioinformatics 24:2098-2100 (10.1093/bioinformatics/btn358)
- Kembel, Cowan, Helmus, Cornwell, Morlon, Ackerly, Blomberg, Webb 2010 Bioinformatics 26(11):1463-1464 (10.1093/bioinformatics/btq166)