library(ggplot2); library(dplyr)
logit <- qlogis; expit <- plogis
te_ink<-"#16241d"; te_body<-"#2c3a31"; te_forest<-"#275139"; te_faint<-"#5d6b61"
te_sage<-"#93a87f"; te_paper<-"#f5f4ee"; te_line<-"#dad9ca"; te_brick<-"#b5534e"; te_gold<-"#cda23f"
theme_te <- function(base_size=12){
theme_minimal(base_size=base_size) +
theme(plot.background=element_rect(fill=te_paper,colour=NA),
panel.background=element_rect(fill="#ffffff",colour=NA),
panel.grid.minor=element_blank(),
panel.grid.major=element_line(colour=te_line,linewidth=0.3),
axis.title=element_text(colour=te_body), axis.text=element_text(colour=te_faint),
plot.title=element_text(colour=te_ink,face="bold"),
plot.subtitle=element_text(colour=te_faint,size=rel(0.9)),
legend.text=element_text(colour=te_faint,size=rel(0.8)))
}Community covariates and species traits
ecology tutorial
R
occupancy
hierarchical models
Bayesian statistics
MCMC
community ecology
Give each species its own slope on a site gradient, and use a trait to explain the variation. A hierarchical model in base R beats a naive two-stage fit.
Species do not all respond to an environmental gradient the same way. Some occupy more sites as the gradient increases; others retreat. A community occupancy model can give every species its own slope on a site covariate, and then ask whether a species trait explains why the slopes differ. That second step is the interesting one: it turns a pile of species-specific responses into a statement about how a trait shapes the response.
The tempting shortcut is to fit each species on its own, collect the slope estimates, and regress them on the trait. That two-stage approach ignores how noisy the per-species slopes are, and the noise is worst for exactly the rare species that carry the least information. This tutorial builds the integrated hierarchical model in base R, and shows it recovering the trait effect where the two-stage fit overstates it.
A gradient, a trait, and species-specific slopes
We simulate 34 species across 95 sites with 4 visits. Occupancy of species s at site r depends on a standardised site covariate through a species-specific intercept and slope. Each species slope is itself drawn around a line in the species trait, so the trait genuinely drives the response.
set.seed(145)
S <- 34L; R <- 95L; K <- 4L
x <- as.numeric(scale(rnorm(R))) # site covariate (standardised)
tr <- as.numeric(scale(rnorm(S))) # species trait (standardised)
mu_a <- logit(0.35); sd_a <- 0.85 # community intercept
beta0 <- 0.20; beta1 <- 0.90; sd_b <- 0.50 # slope depends on trait: b ~ N(beta0 + beta1*tr, sd_b)
mu_p <- logit(0.30); sd_p <- 0.60 # community detection
a_t <- rnorm(S, mu_a, sd_a) # species intercepts
b_t <- rnorm(S, beta0 + beta1*tr, sd_b) # species slopes, tied to the trait
lp_t <- rnorm(S, mu_p, sd_p); p_t <- expit(lp_t)
eta <- matrix(a_t, S, R) + outer(b_t, x); psi <- expit(eta)
z <- matrix(rbinom(S*R, 1, psi), S, R)
y <- matrix(rbinom(S*R, K, ifelse(z==1, p_t, 0)), S, R)
det_sites <- rowSums(y > 0)
c(species=S, sites=R, det_min=min(det_sites), det_max=max(det_sites), total_det=sum(y)) species sites det_min det_max total_det
34 95 6 56 1545 Detection frequencies run from 6 to 56 sites. That range matters: the species at the low end will have slopes that are almost unidentified on their own.
Independent slopes, and why they mislead
Fit each species with its own occupancy-covariate model (as in occupancy with detection covariates), pulling the slope and its standard error from the Hessian.
one <- function(yi){
nll <- function(par){ a<-par[1]; b<-par[2]; p<-expit(par[3]); ps<-expit(a+b*x); d<-yi>0
-(sum(log(ps[d]) + dbinom(yi[d],K,p,log=TRUE)) + sum(log(ps[!d]*(1-p)^K + (1-ps[!d])))) }
op <- try(optim(c(logit(mean(yi>0)+.01),0,0), nll, method="BFGS", hessian=TRUE), silent=TRUE)
if (inherits(op,"try-error")) return(c(b=NA, se=NA))
se <- try(sqrt(diag(solve(op$hessian)))[2], silent=TRUE)
c(b=op$par[2], se=ifelse(inherits(se,"try-error")||is.na(se), NA, se))
}
mle <- t(sapply(1:S, function(s) one(y[s,]))); colnames(mle) <- c("b","se")
round(c(median_SE=median(mle[,"se"]), max_SE=max(mle[,"se"])), 2)median_SE max_SE
0.36 4.82 The median slope has a standard error of 0.36, but the worst case reaches 4.8: one species sits near complete separation on the covariate, and its independent slope diverges to about 11. Now take the tempting shortcut and regress the per-species slopes on the trait.
ok <- is.finite(mle[,"b"]) & is.finite(mle[,"se"]) & mle[,"se"] < 5
naive <- lm(mle[ok,"b"] ~ tr[ok])
c(beta1_naive = round(coef(naive)[2],3), true = beta1)beta1_naive.tr[ok] true
1.482 0.900 The naive two-stage slope is 1.482, well above the true value of 0.9. Regressing point estimates while ignoring their very unequal precision lets the noisiest species drag the trait effect upward.
The integrated hierarchical model
Fit everything at once. Species intercepts come from a community distribution; species slopes come from a line in the trait; detection has its own community distribution. The occupancy state is summed out site by site, and every continuous parameter is updated with random-walk Metropolis.
llmat <- function(a, b, lp){ # per-species log-likelihood, z summed out
e <- matrix(a,S,R) + outer(b,x); ps <- expit(e); p <- expit(lp); Pm <- matrix(p,S,R)
D <- y>0; term <- matrix(0,S,R)
term[D] <- log(ps[D]) + dbinom(y[D],K,Pm[D],log=TRUE)
term[!D] <- log(ps[!D]*(1-Pm[!D])^K + (1-ps[!D]))
rowSums(term)
}
ni <- 16000L; nb <- 6000L; nt <- 2L
a <- rep(logit(0.35),S); b <- rep(0,S); lp <- rep(logit(0.3),S)
m_a<-0; s_a<-1; B0<-0; B1<-0; s_b<-1; m_p<-logit(0.3); s_p<-1
t_a<-0.22; t_b<-0.22; t_lp<-0.22; t_h<-0.13; t_lsd<-0.13
keep <- seq(nb+1,ni,by=nt); nk <- length(keep); kk <- 0L
Bpost <- matrix(NA,nk,S); HYP <- matrix(NA,nk,7) # m_a,s_a,B0,B1,s_b,m_p,s_p
for (it in 1:ni){
ll <- llmat(a,b,lp)
prop <- a + rnorm(S,0,t_a); llp <- llmat(prop,b,lp) # intercepts
au <- log(runif(S)) < (llp+dnorm(prop,m_a,s_a,log=TRUE) - (ll+dnorm(a,m_a,s_a,log=TRUE))); a[au]<-prop[au]
ll <- llmat(a,b,lp)
mb <- B0 + B1*tr
prop <- b + rnorm(S,0,t_b); llp <- llmat(a,prop,lp) # slopes (prior mean B0+B1*trait)
au <- log(runif(S)) < (llp+dnorm(prop,mb,s_b,log=TRUE) - (ll+dnorm(b,mb,s_b,log=TRUE))); b[au]<-prop[au]
ll <- llmat(a,b,lp)
prop <- lp + rnorm(S,0,t_lp); llp <- llmat(a,b,prop) # detection
au <- log(runif(S)) < (llp+dnorm(prop,m_p,s_p,log=TRUE) - (ll+dnorm(lp,m_p,s_p,log=TRUE))); lp[au]<-prop[au]
# hyperparameters
pr<-m_a+rnorm(1,0,t_h); if(log(runif(1))<sum(dnorm(a,pr,s_a,log=TRUE))-sum(dnorm(a,m_a,s_a,log=TRUE)))m_a<-pr
pr<-exp(log(s_a)+rnorm(1,0,t_lsd)); if(log(runif(1))<sum(dnorm(a,m_a,pr,log=TRUE))+dnorm(pr,0,2.5,log=TRUE)+log(pr)-sum(dnorm(a,m_a,s_a,log=TRUE))-dnorm(s_a,0,2.5,log=TRUE)-log(s_a))s_a<-pr
mb<-B0+B1*tr; pr<-B0+rnorm(1,0,t_h); if(log(runif(1))<sum(dnorm(b,pr+B1*tr,s_b,log=TRUE))-sum(dnorm(b,mb,s_b,log=TRUE)))B0<-pr
mb<-B0+B1*tr; pr<-B1+rnorm(1,0,t_h); if(log(runif(1))<sum(dnorm(b,B0+pr*tr,s_b,log=TRUE))-sum(dnorm(b,mb,s_b,log=TRUE)))B1<-pr
mb<-B0+B1*tr; pr<-exp(log(s_b)+rnorm(1,0,t_lsd)); if(log(runif(1))<sum(dnorm(b,mb,pr,log=TRUE))+dnorm(pr,0,2.5,log=TRUE)+log(pr)-sum(dnorm(b,mb,s_b,log=TRUE))-dnorm(s_b,0,2.5,log=TRUE)-log(s_b))s_b<-pr
pr<-m_p+rnorm(1,0,t_h); if(log(runif(1))<sum(dnorm(lp,pr,s_p,log=TRUE))-sum(dnorm(lp,m_p,s_p,log=TRUE)))m_p<-pr
pr<-exp(log(s_p)+rnorm(1,0,t_lsd)); if(log(runif(1))<sum(dnorm(lp,m_p,pr,log=TRUE))+dnorm(pr,0,2.5,log=TRUE)+log(pr)-sum(dnorm(lp,m_p,s_p,log=TRUE))-dnorm(s_p,0,2.5,log=TRUE)-log(s_p))s_p<-pr
if (it>nb && ((it-nb)%%nt==0)){ kk<-kk+1L; Bpost[kk,]<-b; HYP[kk,]<-c(m_a,s_a,B0,B1,s_b,m_p,s_p) }
}
hp <- colMeans(HYP); bhat <- colMeans(Bpost)
round(c(beta0=hp[3], beta1=hp[4], sd_b=hp[5]), 3)beta0 beta1 sd_b
0.288 0.928 0.439 The hierarchical trait effect is 0.928, with a 95 per cent interval from 0.71 to 1.17. That interval covers the true value of 0.9, while the naive two-stage estimate of 1.482 sits above it. The model also recovers the detection spread and the intercept spread reasonably, with a slope standard deviation of 0.44 around the trait line.
Pooling the slopes
The species slopes tell the story directly. Ordered by how often each species was detected, the independent estimates swing widely at the sparse end, with wide error bars; the pooled slopes stay controlled and drift toward the community mean where the data are thin.
df <- data.frame(det=det_sites, mle=mle[,"b"], se=mle[,"se"], hier=bhat)
df <- df[order(df$det),]; df$rank <- 1:S; comm_mean <- mean(bhat)
ggplot(df, aes(rank)) +
geom_hline(yintercept=comm_mean, linetype="dashed", colour=te_faint) +
geom_errorbar(aes(ymin=mle-se, ymax=mle+se), colour=te_brick, width=0, linewidth=0.4, alpha=0.7) +
geom_point(aes(y=mle, shape="Independent (per species) +/-1 SE"), colour=te_brick, size=1.7) +
geom_point(aes(y=hier, shape="Hierarchical (partially pooled)"), colour=te_forest, size=1.7) +
scale_shape_manual(NULL, values=c("Independent (per species) +/-1 SE"=1,"Hierarchical (partially pooled)"=16)) +
coord_cartesian(ylim=c(-4,4)) +
labs(x="species ordered by number of detections (fewest at left)", y="occupancy-covariate slope",
title="Rare and ill-identified slopes are noisy; pooling reins them in",
subtitle="dashed: community mean; one separation case runs off the scale shown") +
theme_te() + theme(legend.position=c(0.72,0.13),
legend.background=element_rect(fill="#ffffff",colour=te_line))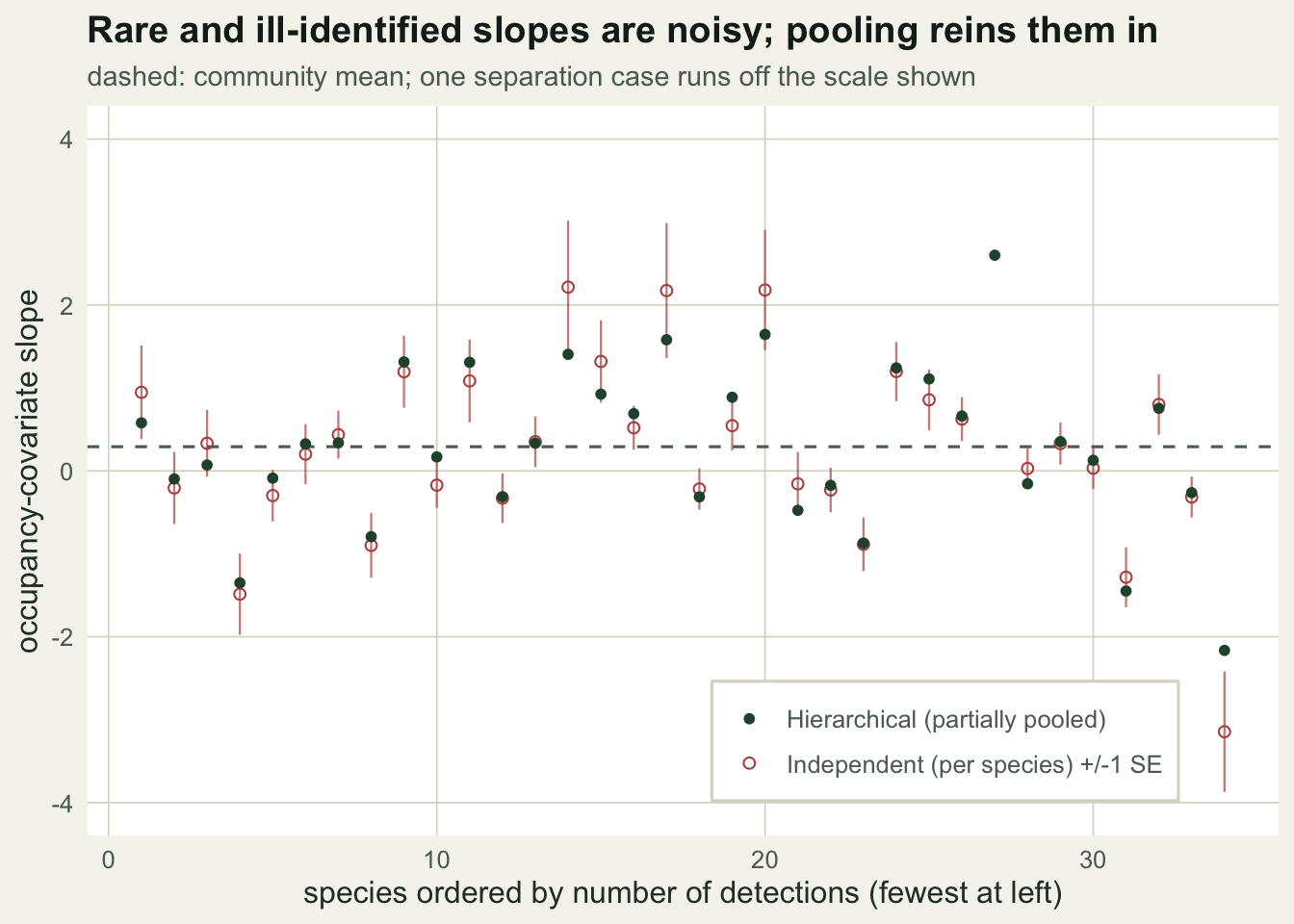
The trait relationship is what the model is really after. Plotting slope against trait, the naive line is visibly steeper than the community relationship the integrated model fits, and the pooled slopes sit close to that line.
b1h <- hp[4]; b0h <- hp[3]; xr <- range(tr)
lh <- data.frame(tr=seq(xr[1],xr[2],length=50)); lh$y <- b0h + b1h*lh$tr
ln <- data.frame(tr=lh$tr, y=coef(naive)[1] + coef(naive)[2]*lh$tr)
dpt <- data.frame(tr=tr, mle=mle[,"b"], hier=bhat)
ggplot() +
geom_point(data=dpt, aes(tr, mle), colour=te_brick, shape=1, size=1.8) +
geom_point(data=dpt, aes(tr, hier), colour=te_forest, size=1.8) +
geom_line(data=ln, aes(tr, y, linetype="Naive two-stage (OLS on MLE slopes)"), colour=te_gold, linewidth=0.8) +
geom_line(data=lh, aes(tr, y, linetype="Hierarchical community relationship"), colour=te_forest, linewidth=0.8) +
scale_linetype_manual(NULL, values=c("Naive two-stage (OLS on MLE slopes)"="dashed","Hierarchical community relationship"="solid")) +
coord_cartesian(ylim=c(-4,4)) +
labs(x="species trait (standardised)", y="occupancy-covariate slope",
title="A trait explains how species respond to the gradient",
subtitle="hollow: independent slopes; filled: pooled; the naive line overstates the trait effect") +
theme_te() + theme(legend.position=c(0.35,0.9),
legend.background=element_rect(fill="#ffffff",colour=te_line))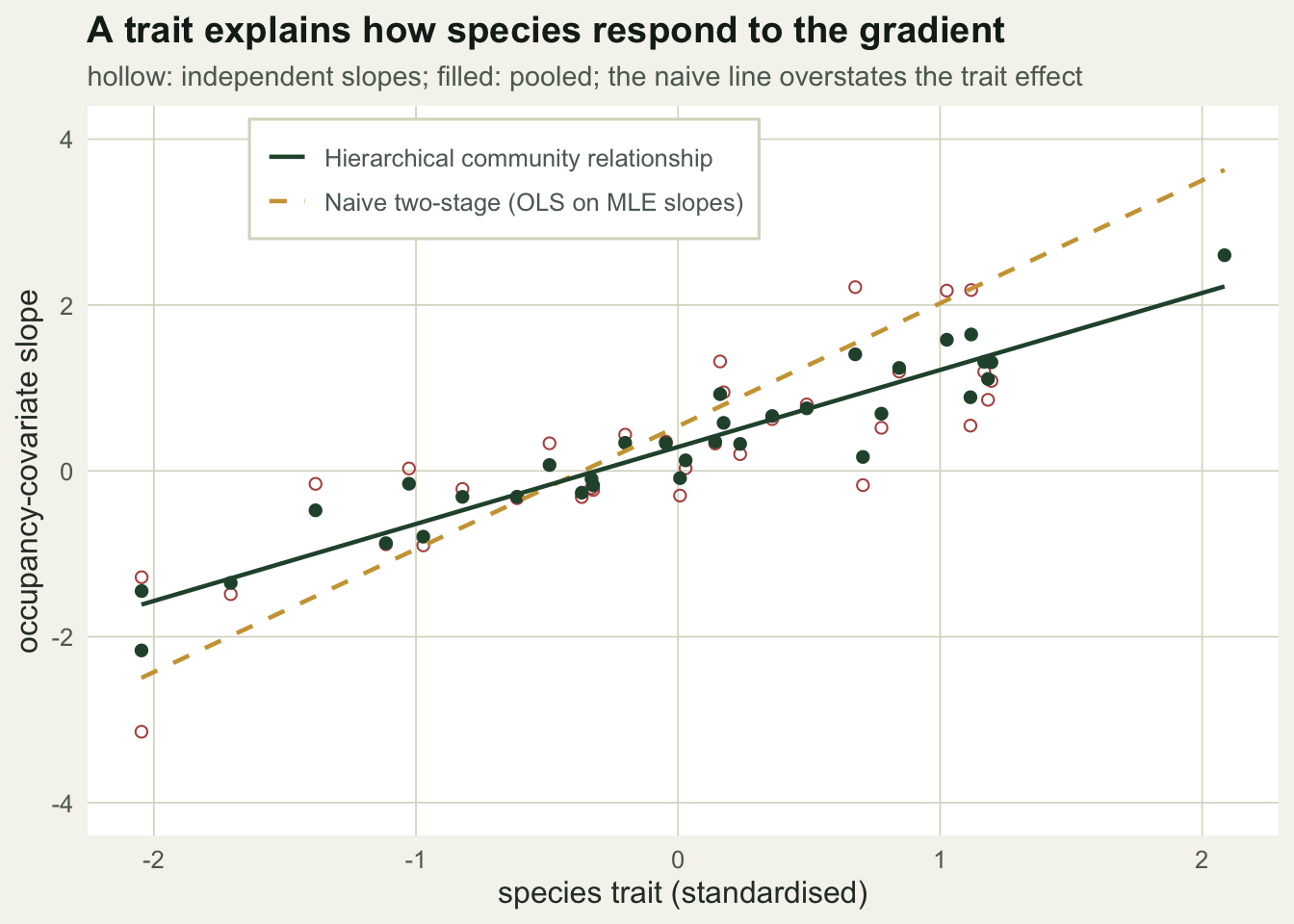
Across all species the gain is plain: the spread of the slope estimates falls from 2.07 for the independent fits to 0.98 for the pooled ones, and the mean distance from the true slopes falls from 0.543 to 0.259, roughly halved.
What to take away
Two-stage analyses are convenient, but regressing noisy estimates on a predictor treats every species as equally informative when the rare ones are barely informative at all. The integrated model weights each species by how much it actually tells us, which is why it recovers the trait effect where the shortcut overstates it. The cost is the same exchangeability assumption as before: species slopes are treated as draws around a common trait line, and if a subgroup breaks that pattern the single relationship will paper over it. Whether the fitted model captures the data it was given is a question for checking a community occupancy model. The starting point, without covariates, is multi-species occupancy models.
References
- Dorazio & Royle 2005 Journal of the American Statistical Association 100(470):389-398 (10.1198/016214505000000015)
- Zipkin, DeWan & Royle 2009 Journal of Applied Ecology 46(4):815-822 (10.1111/j.1365-2664.2009.01664.x)
- Pollock, Tingley, Morris, Golding, O’Hara, Parris, Vesk & McCarthy 2014 Methods in Ecology and Evolution 5(5):397-406 (10.1111/2041-210X.12180)
- Jamil, Ozinga, Kleyer & ter Braak 2013 Journal of Vegetation Science 24(6):988-1000 (10.1111/j.1654-1103.2012.12036.x)
- Ovaskainen, Tikhonov, Norberg, Guillaume Blanchet, Duan, Dunson, Roslin & Abrego 2017 Ecology Letters 20(5):561-576 (10.1111/ele.12757)
- Kery & Royle 2016. Applied Hierarchical Modeling in Ecology, Volume 1. Academic Press. ISBN 978-0-12-801378-6