library(ggplot2)
nll_pois <- function(par, y, K) { # Poisson-abundance marginal NLL
lam <- exp(par[1]); p <- plogis(par[2])
Nv <- 0:K; lp <- dpois(Nv, lam, log = TRUE); ll <- 0
for (i in seq_len(nrow(y))) {
yi <- y[i, ]; idx <- (max(yi) + 1):(K + 1); Ni <- Nv[idx]
lb <- rowSums(vapply(yi, function(v) dbinom(v, Ni, p, log = TRUE),
numeric(length(Ni))))
ll <- ll + log(sum(exp(lp[idx] + lb)))
}
if (!is.finite(ll)) return(1e10)
-ll
}
fit_pois <- function(y, K, start = c(log(2), 0))
optim(start, nll_pois, y = y, K = K, method = "BFGS")The abundance-detection trade-off and N-mixture reliability
R
abundance
imperfect detection
model checking
ecology tutorial
When binomial N-mixture models give unreliable abundance: unbounded estimates, weakly identified negative-binomial dispersion, and the inflation caused by unmodelled detection heterogeneity, shown from scratch in base R.
The binomial N-mixture model looks like a free lunch: point it at repeated counts, no marked animals, and it hands back abundance on an absolute scale. The catch is that the separation of abundance from detection rests entirely on the Poisson and constant-detection assumptions, and the counts alone carry almost no information to check them. When those assumptions bend, the estimate does not merely lose precision; it can run to infinity, hinge on a parameter the data cannot pin down, or double for reasons that have nothing to do with the population. This tutorial works through the three failures that matter most in practice, following the sceptical strand of the literature (Dennis, Morgan and Ridout 2015; Barker et al. 2018; Kery 2018; Link et al. 2018).
The estimator is the same marginal likelihood as before, with the latent abundance summed out. A vectorised version keeps the demonstrations quick:
Failure one: the estimate need not be finite
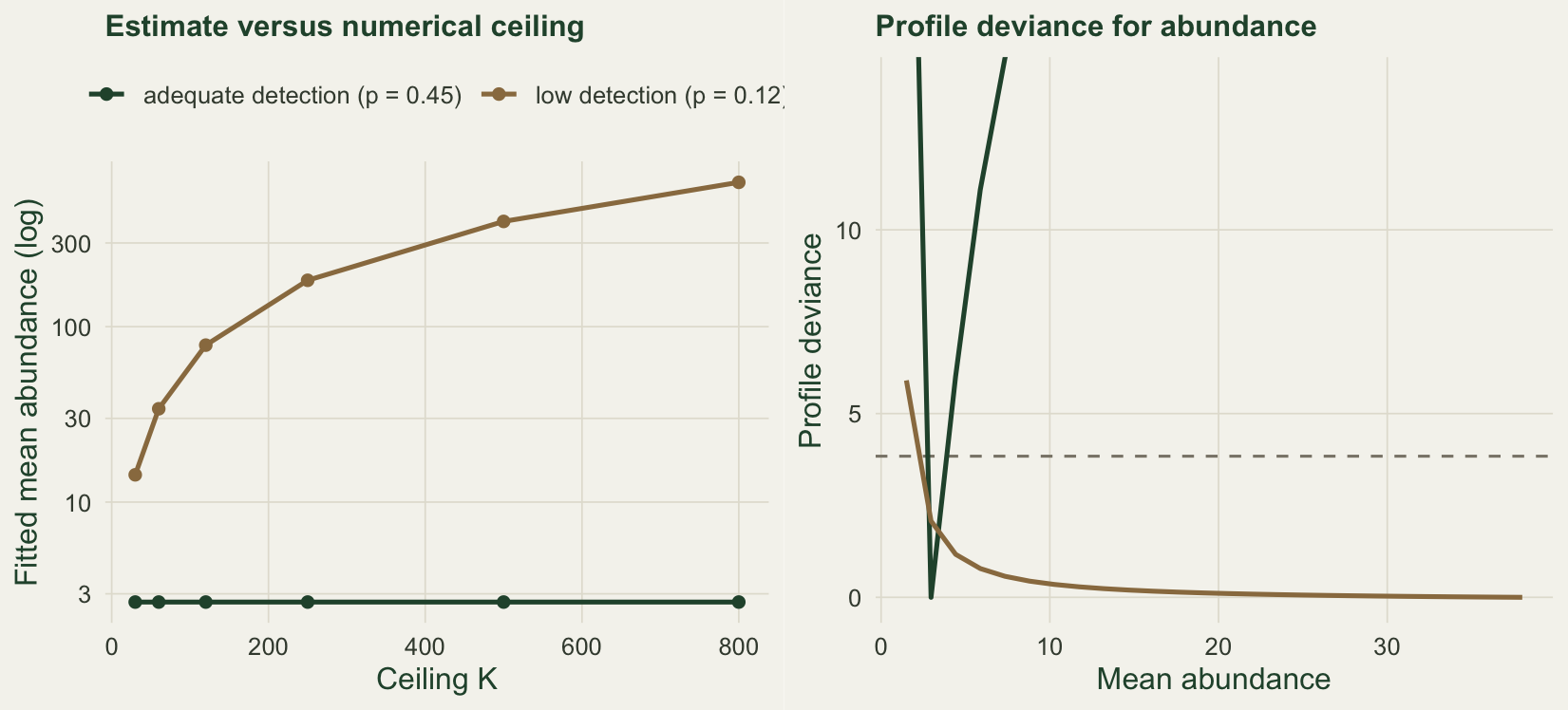
The marginal likelihood does not guarantee an interior maximum. When detection is low and counts are sparse, the likelihood is almost flat along the ridge where the product of abundance and detection matches the mean count, and it keeps improving, by a vanishing amount, as abundance rises and detection falls towards zero. There is then no finite maximum-likelihood estimate: the fitted abundance is pinned to whatever numerical ceiling K you impose, and raising the ceiling simply raises the answer. Dennis, Morgan and Ridout (2015) give the exact condition under which this happens; here we simply watch it occur.
set.seed(4254)
R <- 120L; Tv <- 2L # few visits, low detection
N_lo <- rpois(R, 3.0)
y_lo <- matrix(rbinom(R * Tv, rep(N_lo, Tv), 0.12), R, Tv)
set.seed(4255) # contrast: adequate detection
N_ok <- rpois(R, 3.0)
y_ok <- matrix(rbinom(R * Tv, rep(N_ok, Tv), 0.45), R, Tv)
Kgrid <- c(30, 60, 120, 250, 500, 800)
lam_lo <- sapply(Kgrid, function(K) exp(fit_pois(y_lo, K, c(log(3), qlogis(0.2)))$par[1]))
lam_ok <- sapply(Kgrid, function(K) exp(fit_pois(y_ok, K, c(log(3), 0))$par[1]))
nll_lo30 <- fit_pois(y_lo, 30, c(log(3), qlogis(0.2)))$value
nll_lo800 <- fit_pois(y_lo, 800, c(log(3), qlogis(0.2)))$valueThe low-detection data hold a mean count of only 0.32. Fitting them, the abundance estimate climbs from 14 at a ceiling of 30 to 665 at a ceiling of 800, and it would keep going, while the fit barely changes: the negative log-likelihood moves only from 171.19 to 171.02 across that whole range. The estimate is an artefact of K. Under adequate detection the same procedure is completely stable, returning 2.69 at every ceiling.
Failure two: negative-binomial dispersion is barely identified
The Poisson assumption on abundance is often too rigid, and the natural fix is a negative-binomial mixture with an extra dispersion parameter. Kery (2018) screened well over a hundred bird data sets and found that Poisson and zero-inflated-Poisson N-mixtures were almost always identifiable, whereas negative-binomial versions frequently were not: the dispersion parameter drifts to its boundary and drags the abundance estimate with it. The mechanism is visible even on data that really are Poisson.
nll_nb <- function(par, y, K) { # add a log-dispersion parameter
lam <- exp(par[1]); p <- plogis(par[2]); sz <- exp(par[3])
Nv <- 0:K; lp <- dnbinom(Nv, mu = lam, size = sz, log = TRUE); ll <- 0
for (i in seq_len(nrow(y))) {
yi <- y[i, ]; idx <- (max(yi) + 1):(K + 1); Ni <- Nv[idx]
lb <- rowSums(vapply(yi, function(v) dbinom(v, Ni, p, log = TRUE),
numeric(length(Ni))))
ll <- ll + log(sum(exp(lp[idx] + lb)))
}
if (!is.finite(ll)) return(1e10)
-ll
}
set.seed(4256)
R <- 150L; Tv <- 3L; K <- 80L
N2 <- rpois(R, 2.8)
y2 <- matrix(rbinom(R * Tv, rep(N2, Tv), 0.35), R, Tv) # genuinely Poisson
fp <- fit_pois(y2, K, c(log(2), 0))
fn <- optim(c(log(2), 0, log(5)), nll_nb, y = y2, K = K, method = "BFGS")
aic_p <- 2 * fp$value + 2 * 2
aic_nb <- 2 * fn$value + 2 * 3
nb_size <- exp(fn$par[3])
# how flat is the dispersion? profile the negative log-likelihood at two sizes
prof_sz <- function(s) optim(c(log(2), 0),
function(q) nll_nb(c(q[1], q[2], log(s)), y2, K), method = "Nelder-Mead")$value
nll_s100 <- prof_sz(100); nll_s1000 <- prof_sz(1000)The negative-binomial fit recovers essentially the Poisson answer but wastes a parameter: its dispersion lands at 8472, effectively infinite, which is the Poisson boundary, and its AIC of 1148.3 is worse than the Poisson value of 1146.3. The dispersion is barely identified: increasing it tenfold, from 100 to 1000, changes the negative log-likelihood only from 571.28 to 571.17. On real data with genuine overdispersion the same flatness lets the estimate slide the other way, towards a small dispersion and a large abundance, which is exactly the instability Kery documents. The practical rule that follows is to prefer Poisson or zero-inflated-Poisson mixtures unless overdispersion is both clearly present and, as the next tutorial shows, actually checkable.
Failure three: unmodelled detection heterogeneity inflates abundance
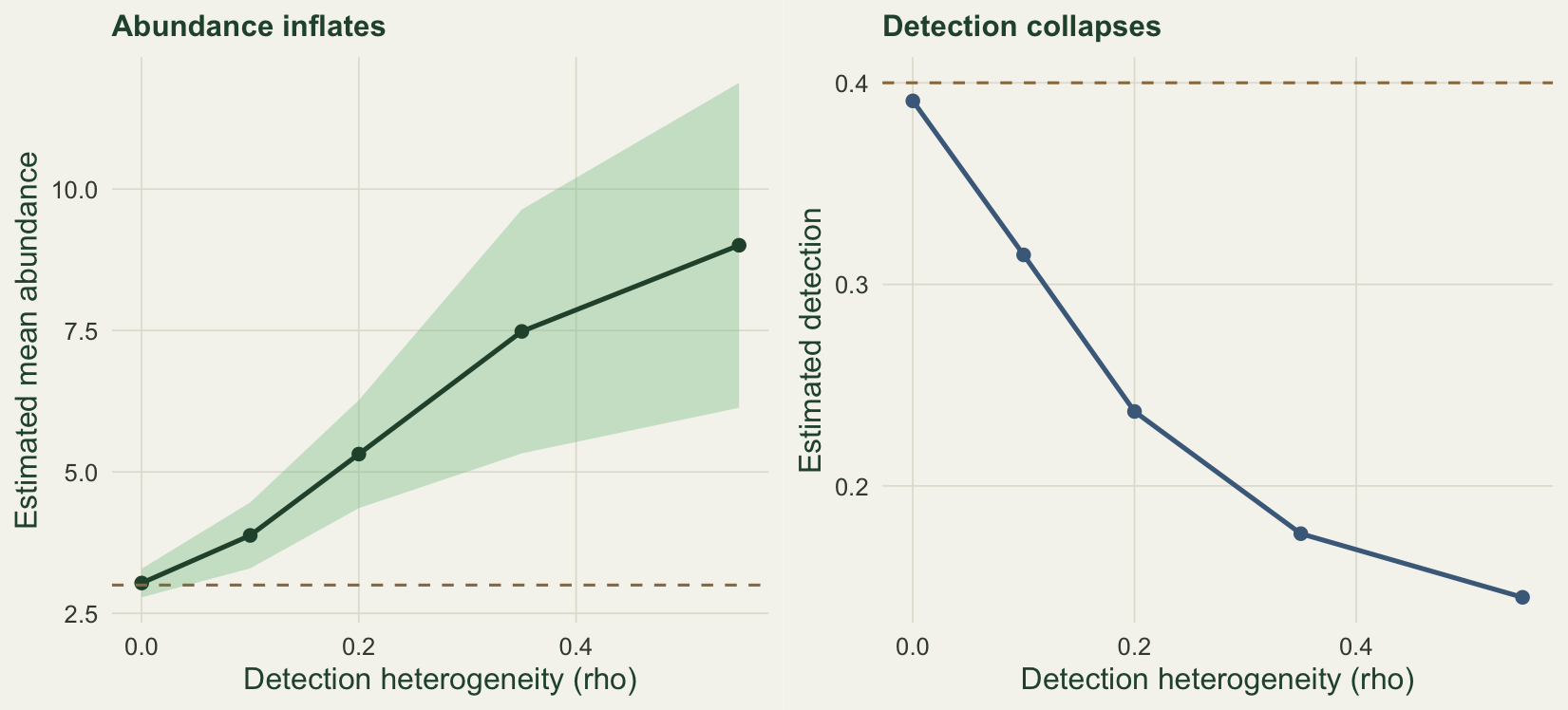
The most dangerous failure is the quietest. The model assumes every individual is detected with the same probability on a given visit, once covariates are accounted for. Real detection varies among individuals, visits and micro-sites in ways no covariate captures. Barker et al. (2018) and Link et al. (2018) showed that even small departures from constant detection can bias abundance badly, and that the counts give little warning. The reason is mechanical: extra variation in the counts looks, to the model, like extra variation in abundance, so it raises the mean abundance to explain the spread. We can watch the inflation grow by adding beta-binomial variation to detection and increasing it.
# beta-binomial counts: detection probability itself varies, with intra-visit
# correlation rho; rho = 0 is the ordinary binomial, larger rho is more heterogeneity
rbb <- function(N, p, rho) {
a <- p * (1 - rho) / rho; b <- (1 - p) * (1 - rho) / rho
rbinom(length(N), N, rbeta(length(N), a, b))
}
set.seed(4257)
R <- 150L; Tv <- 4L; K <- 55L; reps <- 8L; lam_true <- 3.0; p_true <- 0.40
rhos <- c(0.0001, 0.10, 0.20, 0.35, 0.55)
sweep <- do.call(rbind, lapply(rhos, function(rh) {
m <- t(replicate(reps, {
N <- rpois(R, lam_true); y <- sapply(1:Tv, function(t) rbb(N, p_true, rh))
f <- fit_pois(y, K, c(log(3), qlogis(0.4))); c(exp(f$par[1]), plogis(f$par[2]))
}))
data.frame(rho = rh, lam = mean(m[, 1]), lam_sd = sd(m[, 1]), p = mean(m[, 2]))
}))With no heterogeneity the fit is honest, recovering a mean abundance of 3.04 against the truth of 3.0. As detection heterogeneity rises, abundance inflates steadily, reaching 9.01 at the strongest level, more than 200 per cent above the truth, while the estimated detection collapses from 0.39 towards 0.14. Nothing in the counts announces this. The model reports tight confidence intervals around a badly wrong number, which is worse than an honestly wide one.
When the model can be trusted
None of this makes N-mixture models useless. It marks out where they are safe. A Poisson or zero-inflated-Poisson mixture, fitted to data with reasonably high detection and genuine population closure across visits, and checked for fit, gives defensible abundance. The trouble arrives with low detection, with negative-binomial dispersion added on faith, and above all with detection heterogeneity that no covariate removes. Barker et al. (2018) and Link et al. (2018) draw the blunt conclusion that when an absolute abundance really matters, the counts should be supplemented with data that identify detection directly, such as recaptures of marked individuals, rather than asked to do the job alone.
That leaves a practical question: given a single fitted model, how do you tell which regime you are in? The companion tutorial on checking an N-mixture model builds the diagnostics, from bootstrap goodness-of-fit and an overdispersion ratio to residual plots and the sensitivity to K we met above.
References
Dennis, E.B., Morgan, B.J.T. and Ridout, M.S. 2015. Biometrics 71(1):237-246 (10.1111/biom.12246).
Barker, R.J., Schofield, M.R., Link, W.A. and Sauer, J.R. 2018. Biometrics 74(1):369-377 (10.1111/biom.12734).
Kery, M. 2018. Ecology 99(2):281-288 (10.1002/ecy.2093).
Link, W.A., Schofield, M.R., Barker, R.J. and Sauer, J.R. 2018. Ecology 99(7):1547-1551 (10.1002/ecy.2362).
Royle, J.A. 2004. Biometrics 60(1):108-115 (10.1111/j.0006-341X.2004.00142.x).