library(ape)
library(nlme)
library(ggplot2)
te_paper <- "#f5f4ee"; te_ink <- "#16241d"; te_body <- "#2c3a31"
te_faint <- "#5d6b61"; te_sage <- "#93a87f"; te_rust <- "#b5534e"
te_line <- "#dad9ca"; te_low <- "#c9b458"; te_high <- "#1d5b4e"
theme_te <- function(base_size = 12) {
theme_minimal(base_size = base_size) +
theme(
plot.background = element_rect(fill = te_paper, colour = NA),
panel.background = element_rect(fill = te_paper, colour = NA),
panel.grid.major = element_line(colour = te_line, linewidth = 0.3),
panel.grid.minor = element_blank(),
axis.text = element_text(colour = te_body),
axis.title = element_text(colour = te_ink),
plot.title = element_text(colour = te_ink, face = "bold"),
strip.text = element_text(colour = te_ink, face = "bold")
)
}Phylogenetic signal: Blomberg’s K and Pagel’s lambda
R
ape
nlme
phylogenetics
comparative methods
ecology tutorial
Measure phylogenetic signal in species traits with Blomberg’s K and Pagel’s lambda in R: fit both indices, test them against a null, and read the outcome.
Closely related species tend to resemble one another, because they share ancestry and have had less time to diverge. Before you run any comparative analysis it helps to ask a prior question: does a trait carry phylogenetic signal at all, and how strong is it? Two indices dominate the literature, and you can compute both in R with ape and nlme alone.
Pagel’s lambda rescales the internal branches of the tree so that the covariance implied by the phylogeny best matches the trait. Lambda near 1 means the trait evolves as if by Brownian motion along the tree; lambda near 0 means the phylogeny carries no information (a star, where every species is independent). Blomberg’s K compares the variance among species to the variance expected under Brownian motion on that exact tree. K near 1 is Brownian-strength signal, K below 1 is weaker than Brownian, and K above 1 means relatives are even more alike than Brownian motion predicts.
Setup
A tree and two traits
There is no field dataset here: the point is to see each index behave against a trait whose history we control. We simulate one ultrametric tree of 40 species, then evolve two traits on it. The first, standing in for something like log body mass, evolves by Brownian motion (Pagel’s lambda set to 1). The second is a labile trait with only a trace of structure (lambda set to 0.1), the kind of behaviour or habitat preference that turns over quickly.
set.seed(414)
tree <- rcoal(40)
tree$tip.label <- sprintf("sp%02d", 1:40)
# simulate a trait with a given Pagel's lambda: scale off-diagonal covariances
sim_pagel <- function(phy, lambda, sigma2 = 1) {
V <- vcv(phy); Vl <- V * lambda; diag(Vl) <- diag(V)
x <- MASS::mvrnorm(1, mu = rep(0, nrow(V)), Sigma = sigma2 * Vl)
setNames(x, rownames(V))
}
set.seed(2) ; body_mass <- sim_pagel(tree, lambda = 1.0)
set.seed(291) ; labile_trait <- sim_pagel(tree, lambda = 0.1)Blomberg’s K by hand
K has a closed form, so there is no need for an extra package. The recipe: find the phylogenetic mean (a generalised-least-squares mean that weights species by the inverse of the phylogenetic covariance), then take the ratio of the raw variance around that mean to the phylogeny-corrected variance, and divide by the value that ratio would take under Brownian motion.
blomberg_K <- function(x, phy) {
x <- x[phy$tip.label]; n <- length(x)
V <- vcv(phy); Vi <- solve(V); one <- rep(1, n)
a <- as.numeric((t(one) %*% Vi %*% x) / (t(one) %*% Vi %*% one)) # phylo mean
d <- x - a
mse_star <- as.numeric(t(d) %*% d) / (n - 1) # ignores the tree
mse_phy <- as.numeric(t(d) %*% Vi %*% d) / (n - 1) # corrects for the tree
expected <- (sum(diag(V)) - n / sum(Vi)) / (n - 1) # value under Brownian motion
(mse_star / mse_phy) / expected
}
K_mass <- blomberg_K(body_mass, tree)
K_labile <- blomberg_K(labile_trait, tree)
c(body_mass = K_mass, labile = K_labile)body_mass labile
1.1024212 0.0102723 The point estimate on its own does not tell you whether signal is more than you would see by chance. The standard test shuffles trait values across the tips many times and rebuilds the null distribution of K; a value in the far right tail means the observed clustering is unlikely without shared history.
set.seed(303); K_mass_null <- replicate(999, blomberg_K(setNames(sample(body_mass), tree$tip.label), tree))
set.seed(304); K_labile_null <- replicate(999, blomberg_K(setNames(sample(labile_trait), tree$tip.label), tree))
p_mass <- (1 + sum(K_mass_null >= K_mass)) / 1000
p_labile <- (1 + sum(K_labile_null >= K_labile)) / 1000
c(p_mass = p_mass, p_labile = p_labile) p_mass p_labile
0.001 0.555 Body mass gives K of 1.1 with a randomisation p of 0.001; the labile trait gives K of 0.01 with p of 0.56. The first is Brownian-strength signal and highly unlikely by chance; the second is indistinguishable from a phylogeny-free arrangement.
Pagel’s lambda by maximum likelihood
Lambda has a natural likelihood estimate. Fit an intercept-only generalised least squares model with a corPagel correlation structure and let lambda vary; the fitted lambda is the maximum-likelihood value. To test it, compare against the model with lambda fixed at 0, which for an ultrametric tree is exactly the ordinary model with independent, equal-variance errors (a star phylogeny). Twice the difference in log-likelihood is a likelihood-ratio statistic on one degree of freedom.
est_lambda <- function(x, phy) {
d <- data.frame(sp = phy$tip.label, y = x[phy$tip.label])
m1 <- gls(y ~ 1, data = d, correlation = corPagel(0.3, phy, form = ~sp), method = "ML")
m0 <- gls(y ~ 1, data = d, method = "ML") # lambda = 0 (star)
lam <- as.numeric(coef(m1$modelStruct$corStruct, unconstrained = FALSE))
lr <- as.numeric(2 * (logLik(m1) - logLik(m0)))
c(lambda = lam, LR = lr, p = pchisq(lr, df = 1, lower.tail = FALSE))
}
round(est_lambda(body_mass, tree), 4) lambda LR p
1.0005 82.2366 0.0000 round(est_lambda(labile_trait, tree), 4)lambda LR p
0.2156 1.1874 0.2759 Body mass returns lambda of essentially 1 with a likelihood-ratio test that is decisive, matching the K result. The labile trait returns lambda near 0.2, and the likelihood-ratio test does not reject lambda of 0: there is no signal worth acting on. The two indices need not agree to the decimal, because K summarises a variance ratio and lambda summarises a branch-scaling; here they agree on the verdict, which is what matters.
Seeing it on the tree
ramp <- colorRampPalette(c(te_low, te_high))
col_from <- function(v) ramp(100)[as.integer(cut(v, breaks = seq(min(v), max(v), length.out = 101), include.lowest = TRUE))]
op <- par(mfrow = c(1, 2), mar = c(1, 0.5, 2.5, 0.5), bg = te_paper, fg = te_body, col.main = te_ink)
plot(tree, show.tip.label = FALSE, edge.color = te_faint, edge.width = 1.2)
tiplabels(pch = 21, bg = col_from(body_mass[tree$tip.label]), col = te_ink, cex = 1.1)
title(main = sprintf("body mass (K = %.2f, p = %.3f)", K_mass, p_mass), cex.main = 0.95, font.main = 1)
plot(tree, show.tip.label = FALSE, edge.color = te_faint, edge.width = 1.2)
tiplabels(pch = 21, bg = col_from(labile_trait[tree$tip.label]), col = te_ink, cex = 1.1)
title(main = sprintf("labile trait (K = %.2f, p = %.2f)", K_labile, p_labile), cex.main = 0.95, font.main = 1)
par(op)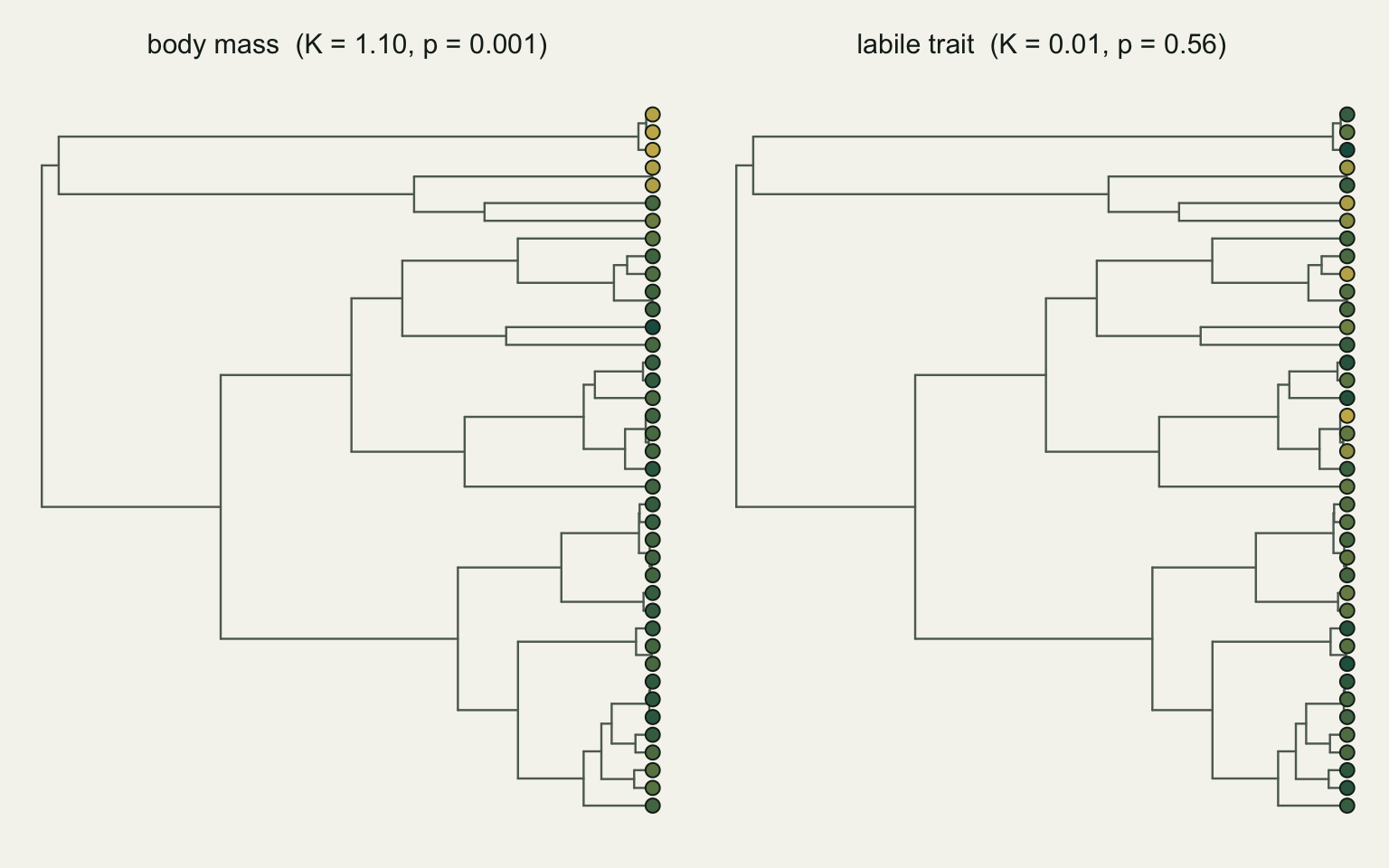
dfn <- rbind(data.frame(trait = "body mass", K = K_mass_null, obs = K_mass),
data.frame(trait = "labile trait", K = K_labile_null, obs = K_labile))
dfn$trait <- factor(dfn$trait, levels = c("body mass", "labile trait"))
ggplot(dfn, aes(K)) +
geom_histogram(bins = 40, fill = te_sage, colour = te_paper, linewidth = 0.2) +
geom_vline(aes(xintercept = obs), colour = te_rust, linewidth = 1) +
facet_wrap(~trait, scales = "free_x") +
labs(x = "Blomberg's K under tip randomisation", y = "null replicates",
title = "Observed K (red) against a phylogeny-free null") +
theme_te(12)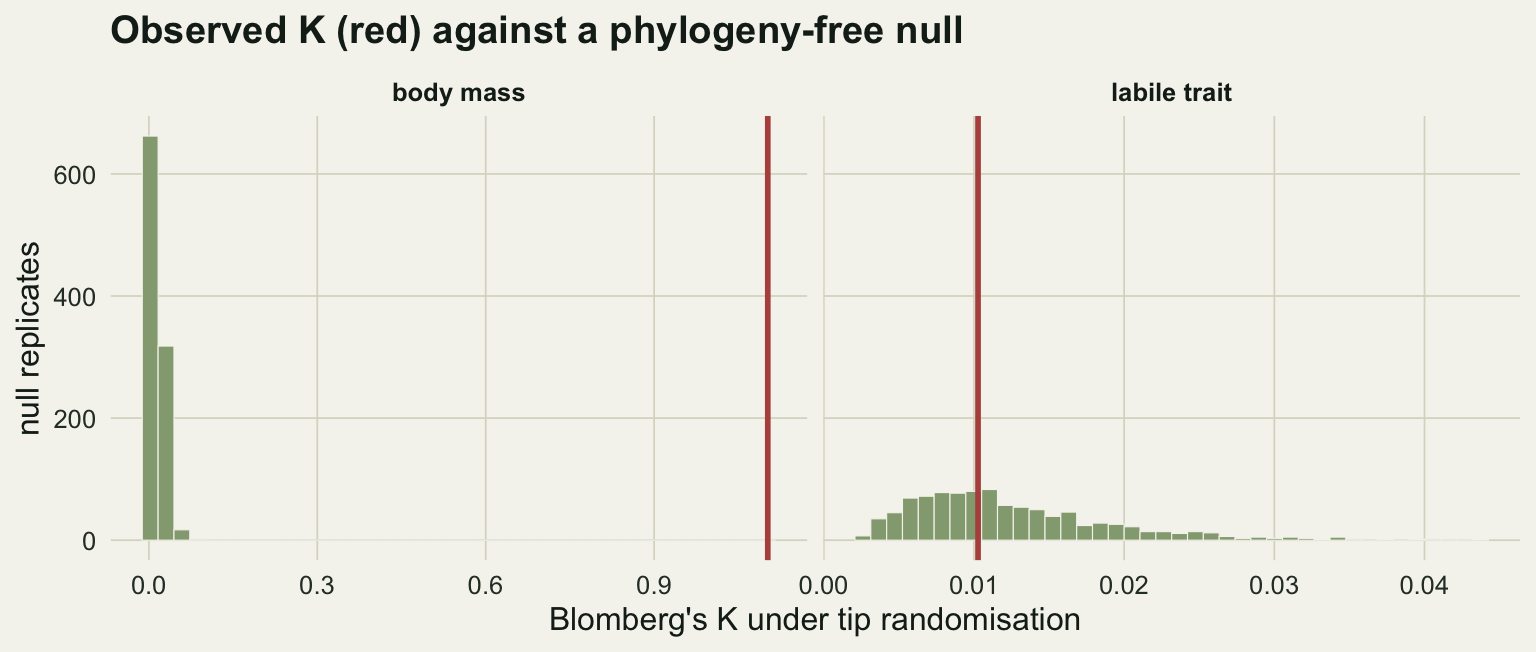
Reading the two indices
Lambda and K answer slightly different questions, and it is worth keeping them apart. Lambda asks how much the branch lengths need to shrink for the phylogeny to explain the trait covariance, and it lives on a natural 0 to 1 scale that many people find easy to report. K asks how the observed variance among species compares to a Brownian expectation, so it can exceed 1 when close relatives are more alike than drift alone would produce, for example under strong niche conservatism. A trait can show a moderate lambda yet a small K, or the reverse, and neither is wrong; they are different summaries.
A few cautions. The point estimate of K from a single trait is noisy: draw a Brownian trait on the same tree a thousand times and K scatters widely around 1, so lean on the test rather than the exact value. Both indices assume the tree and its branch lengths are correct, and error in either propagates into the estimate. Most importantly, signal is a pattern, not a process. A high lambda is consistent with Brownian drift, but also with slow stabilising selection or shared environments among relatives; the index tells you the trait is phylogenetically structured, not why.
If a trait shows signal, the species are not independent data points, and analyses that ignore that will misbehave. That is the starting point for phylogenetic generalised least squares and independent contrasts.
References
- Pagel 1999 Nature 401:877-884 (10.1038/44766)
- Blomberg, Garland & Ives 2003 Evolution 57(4):717-745 (10.1111/j.0014-3820.2003.tb00285.x)
- Freckleton, Harvey & Pagel 2002 The American Naturalist 160(6):712-726 (10.1086/343873)
- Revell, Harmon & Collar 2008 Systematic Biology 57(4):591-601 (10.1080/10635150802302427)
- Munkemuller et al. 2012 Methods in Ecology and Evolution 3(4):743-756 (10.1111/j.2041-210X.2012.00196.x)
- Paradis & Schliep 2019 Bioinformatics 35(3):526-528 (10.1093/bioinformatics/bty633)