library(vegan)
library(dplyr)
library(ggplot2)
set.seed(11)
S <- 110
geom_sad <- function(R, k) { w <- k^(0:(R - 1)); w / sum(w) } # uneven
even_sad <- function(R) rep(1 / R, R) # even
draw_site <- function(rich, N, sad) {
cnt <- as.vector(rmultinom(1, N, sad))
out <- numeric(S); out[seq_len(rich)] <- cnt; out
}
comm <- rbind(
Riverine = draw_site(95, 6000, geom_sad(95, 0.88)), # rich, uneven, deep sample
Upland = draw_site(52, 170, even_sad(52)), # even, shallow sample
Disturbed = draw_site(16, 600, geom_sad(16, 0.80)) # genuinely poor
)
colnames(comm) <- paste0("sp", seq_len(S))Rarefaction and accumulation curves in R: comparing richness when effort differs
community ecology
diversity
R
More individuals and more plots both mean more species observed, whatever the true richness. Rarefaction and species accumulation curves separate real diversity from sampling effort, with vegan in R.
Species richness, the plain count of species in a sample, is the most intuitive diversity measure there is. It is also the most easily fooled by effort. Count more individuals and you find more species. Walk more plots and you find more species. Neither increase tells you the community is richer; it tells you that you looked harder. When two sites were sampled with different effort, their raw species counts are not comparable, and ranking them by those counts can be exactly wrong.
Rarefaction and accumulation curves are the standard fix. They put richness on a common footing so that a comparison reflects the communities rather than the fieldwork. This post builds both in R with the vegan package and shows a case where the raw ranking reverses once effort is equalized.
Two questions, two curves
The two curve types answer different questions, and mixing them up is a common source of confusion.
Individual-based rarefaction asks: if I had counted only n individuals at this site, how many species would I expect to have seen? It interpolates a single site’s richness down to smaller numbers of individuals, using the formula from Hurlbert (1971). This is the tool for comparing sites that were sampled to different depths: read every site’s curve down to the smallest site’s sample size and compare there.
Sample-based accumulation asks: as I add survey plots, how does the total species count grow? It accumulates richness across sampling units rather than individuals. This is the tool for a different question, whether a survey has captured most of the species present, or whether more plots would keep turning up new ones.
The first is about fair comparison; the second is about completeness. Both come from the same idea, that richness is a function of effort and you have to say which level of effort you mean.
The trap: comparing raw counts
Gotelli and Colwell (2001) laid out why raw richness comparisons mislead and why rarefaction is the remedy. The short version: a larger sample almost always contains more species than a smaller one drawn from the same community, purely because rarer species need more draws to appear. So a site that was sampled harder gets a head start that has nothing to do with its diversity. The worked example below makes that head start visible, and shows it can be large enough to flip a ranking.
Worked example 1: individual-based rarefaction
Here are three sites built so that sampling effort and true richness pull in different directions. The data is synthetic and illustrative. “Riverine” is a rich but uneven community sampled very deeply (6000 individuals). “Upland” is an even community sampled shallowly (170 individuals). “Disturbed” is a genuinely species-poor community sampled moderately.
First the raw and rarefied richness side by side. Observed richness is just the count of species present; rarefy gives the expected richness if each site were thinned to the common sample size, here 170 individuals, the size of the smallest site.
common_N <- min(rowSums(comm))
data.frame(
individuals = rowSums(comm),
observed = specnumber(comm),
rarefied = round(as.numeric(rarefy(comm, common_N)), 1)
) individuals observed rarefied
Riverine 6000 61 28.4
Upland 170 51 51.0
Disturbed 600 16 14.7Read those two richness columns against each other. By raw count, Riverine looks like the richest site, ten species ahead of Upland. Thin everything to 170 individuals and the order reverses: Upland is expected to hold about 51 species at that effort, while Riverine drops to roughly 28. Riverine’s apparent lead was an artefact of counting 6000 individuals instead of 170. Disturbed, meanwhile, stays poor whether you rarefy it or not, which is the other half of the lesson: its low count is real, not a sampling shortfall.
The curves show why. Plotting expected richness against number of individuals for each site, the comparison is whatever sits above the dashed common-effort line.
curve_df <- do.call(rbind, lapply(rownames(comm), function(s) {
grid <- unique(round(c(seq(1, sum(comm[s, ]), length.out = 60), sum(comm[s, ]))))
data.frame(site = s, n = grid,
richness = as.numeric(rarefy(comm[s, , drop = FALSE], grid)))
}))
rare_at_common <- data.frame(site = rownames(comm), n = common_N,
richness = as.numeric(rarefy(comm, common_N)))
endpts <- curve_df %>% group_by(site) %>% slice_max(n, n = 1) %>% ungroup()
sitecols <- c(Riverine = "#275139", Upland = "#c98a2e", Disturbed = "#3a7ca5")
ggplot(curve_df, aes(n, richness, color = site)) +
geom_vline(xintercept = common_N, linetype = "dashed", color = "#16241d", linewidth = 0.4) +
geom_line(linewidth = 0.9) +
geom_point(data = endpts, size = 2.6) +
geom_point(data = rare_at_common, shape = 21, fill = "white", stroke = 1.1, size = 2.6) +
annotate("text", x = common_N, y = 2, label = paste0("common N = ", common_N),
hjust = -0.05, size = 3.2, color = "#16241d") +
scale_color_manual(values = sitecols, name = NULL) +
labs(x = "individuals sampled", y = "expected species richness") +
theme_minimal(base_size = 12) + theme(legend.position = "top")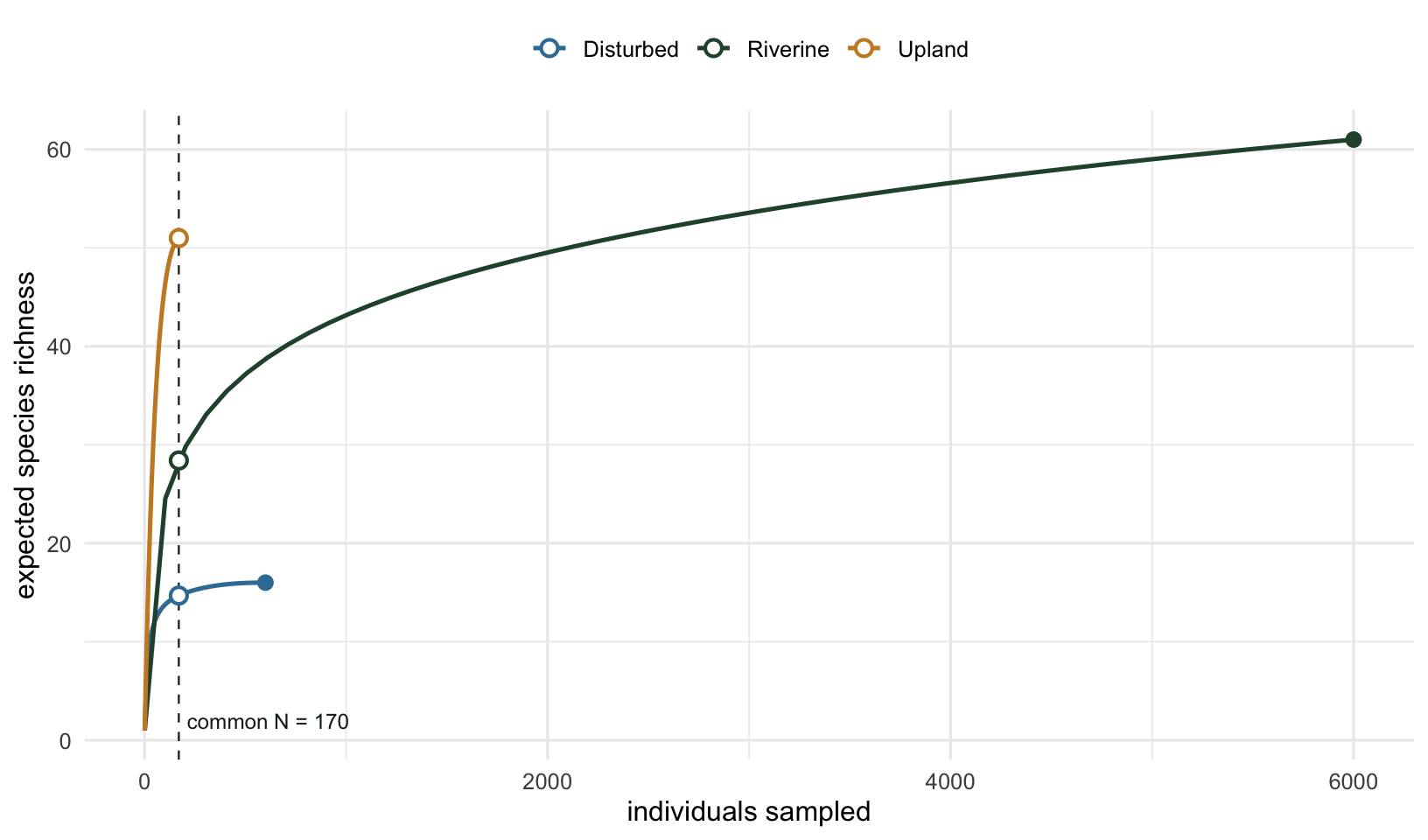
The filled dots are each site’s observed richness at its own sample size, the open dots are the rarefied values at the common effort. Upland’s curve climbs fast because its species are evenly abundant, so a handful of individuals already turns up most of them. Riverine’s climbs slowly because a few common species dominate the early draws and the rest are rare, so it takes thousands of individuals to accumulate the same richness. The deep sample lets Riverine eventually pass Upland in raw count, but at equal effort it is nowhere close. The vertical gap at the dashed line is the honest comparison.
Worked example 2: accumulation and how much is left
The second question is completeness. Here is a survey of 16 plots from one habitat, and the species accumulation curve as plots are added. specaccum with the exact method gives the expected curve and its standard deviation analytically, and specpool estimates how many species the habitat holds in total, including ones not yet seen.
set.seed(21)
n_plots <- 16; Spool <- 40
commonness <- sort(rbeta(Spool, 0.7, 1.3), decreasing = TRUE)
plots <- t(sapply(seq_len(n_plots), function(p) rbinom(Spool, 1, commonness) * rpois(Spool, 6)))
colnames(plots) <- paste0("sp", seq_len(Spool))
plots <- plots[, colSums(plots) > 0, drop = FALSE]
sac <- specaccum(plots, method = "exact")
pool <- specpool(plots)
round(pool[c("Species", "chao", "chao.se", "jack1")], 1) Species chao chao.se jack1
All 34 39.9 7.1 38.7The survey observed 34 species across the 16 plots. The Chao2 estimator puts the total closer to 40, so roughly 85% of the estimated species pool has been found, and a handful are likely still out there. The curve below makes the gap concrete: it has bent over but not flattened, and the distance up to the Chao2 line is the richness more plots would probably recover.
sac_df <- data.frame(sites = sac$sites, richness = sac$richness, sd = sac$sd)
ggplot(sac_df, aes(sites, richness)) +
geom_ribbon(aes(ymin = richness - 2 * sd, ymax = richness + 2 * sd),
fill = "#275139", alpha = 0.15) +
geom_line(color = "#275139", linewidth = 0.9) +
geom_point(color = "#275139", size = 2) +
geom_hline(yintercept = pool$chao, linetype = "dashed", color = "#c98a2e", linewidth = 0.5) +
annotate("text", x = 2, y = pool$chao, label = paste0("Chao2 estimate = ", round(pool$chao)),
vjust = -0.6, hjust = 0, size = 3.2, color = "#c98a2e") +
scale_x_continuous(breaks = seq(2, 16, 2)) +
labs(x = "plots surveyed", y = "accumulated species richness") +
theme_minimal(base_size = 12)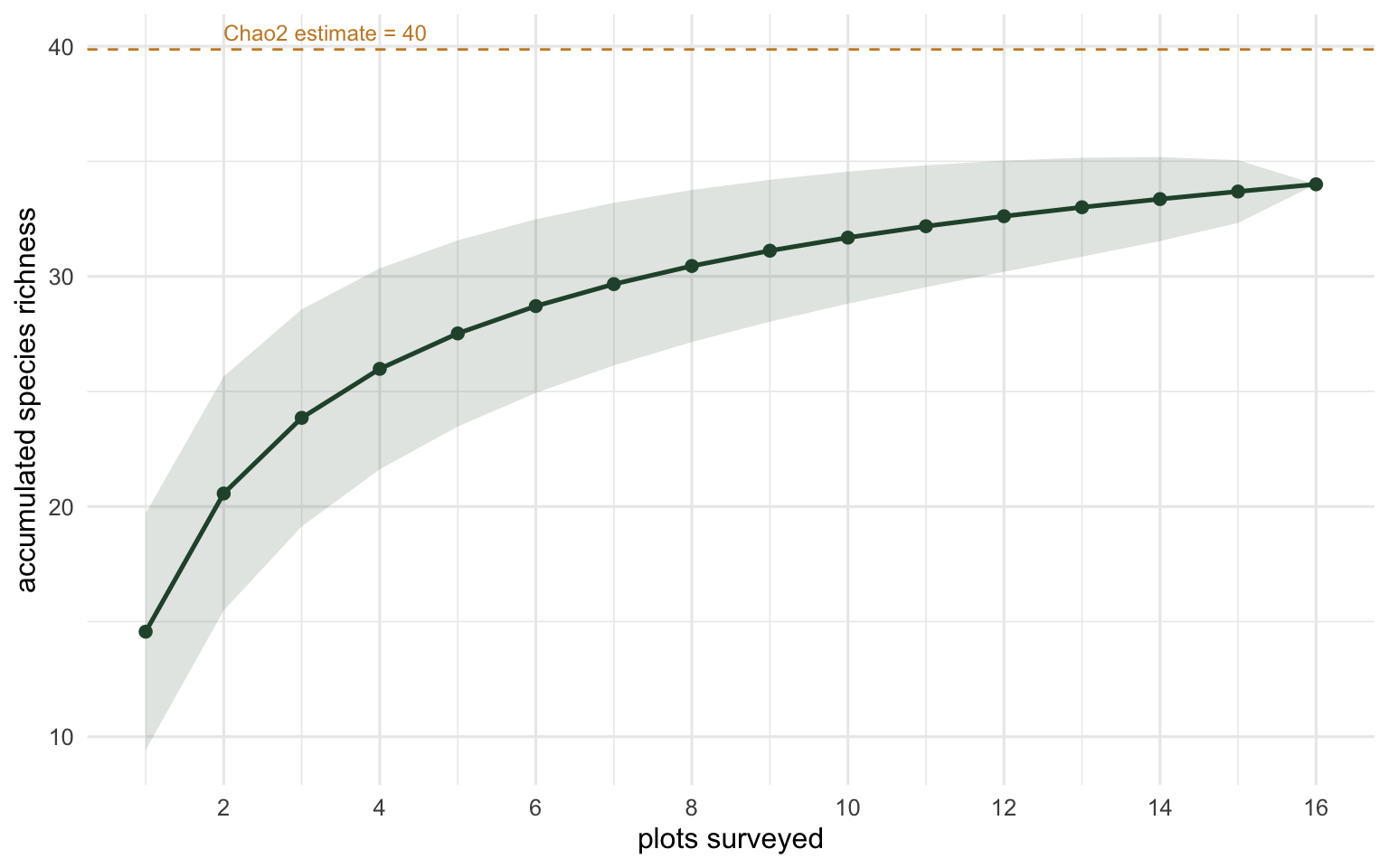
A curve that has plateaued says the survey is close to complete; one still climbing steeply says keep sampling. The Chao2 line turns that visual impression into a number you can report.
The modern framing: Hill numbers and coverage
The approach here, rarefy everything to a common number of individuals or plots, is the classic one and still perfectly serviceable. Two refinements have become standard since. First, Colwell and colleagues (2012) unified rarefaction with extrapolation, so you can project a curve a little beyond your sample rather than only thinning the larger ones down, which avoids discarding data from the deep samples. Second, Chao and Jost (2012) argued for standardizing by sample completeness (coverage) instead of raw size, since equal numbers of individuals can still represent very different fractions of two communities. Both ideas, together with Hill numbers as a richness-to-evenness family, are implemented in the iNEXT package (Hsieh, Ma and Chao 2016). For a serious diversity comparison today, that is the toolkit to reach for; the vegan functions shown here are the right way to understand what it is doing underneath.
Caveats
Rarefaction assumes your counts are counts of individuals drawn from the community, and that the samples are comparable in everything except size. If individuals are not independent (clustered colonial organisms, or counts that are really biomass), the expected-richness formula does not strictly apply.
Down-sampling to the smallest site throws away information from the larger samples. With very unequal sample sizes, the common effort can be so small that the comparison is imprecise, which is part of why extrapolation-based methods became popular.
Richness, the focus here, is the diversity measure most sensitive to rare species and therefore to sampling. If you care about the common, abundant part of the community, an evenness-weighted measure (Shannon, Simpson, or Hill numbers of order one or two) is both more stable and often more ecologically relevant. Richness is where rarefaction matters most precisely because it is the most effort-dependent.
References
Chao, A., and Jost, L. (2012). Coverage-based rarefaction and extrapolation: standardizing samples by completeness rather than size. Ecology, 93, 2533-2547.
Colwell, R.K., Chao, A., Gotelli, N.J., Lin, S.-Y., Mao, C.X., Chazdon, R.L., and Longino, J.T. (2012). Models and estimators linking individual-based and sample-based rarefaction, extrapolation and comparison of assemblages. Journal of Plant Ecology, 5, 3-21.
Gotelli, N.J., and Colwell, R.K. (2001). Quantifying biodiversity: procedures and pitfalls in the measurement and comparison of species richness. Ecology Letters, 4, 379-391.
Hsieh, T.C., Ma, K.H., and Chao, A. (2016). iNEXT: an R package for rarefaction and extrapolation of species diversity (Hill numbers). Methods in Ecology and Evolution, 7, 1451-1456.
Hurlbert, S.H. (1971). The nonconcept of species diversity: a critique and alternative parameters. Ecology, 52, 577-586.