library(vegan)
set.seed(37)
S <- 24; n <- 60
x1 <- runif(n, -1, 1); x2 <- runif(n, -1, 1)
opt1 <- seq(-1.2, 1.2, length.out = S)
opt2 <- sample(seq(-1.2, 1.2, length.out = S))
lambda <- sapply(1:S, function(j)
18 * exp(-0.5*((x1 - opt1[j])/0.6)^2 - 0.5*((x2 - opt2[j])/0.7)^2))
comm <- matrix(rpois(n * S, lambda), n, S)
comm[rowSums(comm) == 0, 1] <- 1
env <- data.frame(x1, x2)
D <- vegdist(comm, "bray")capscale and dbrda: the same ordination, a different test
R
ordination
vegan
multivariate
vegan has two functions for distance-based constrained ordination. On a Bray-Curtis dissimilarity they return the same axes but a different pseudo-F, and the reason is the negative eigenvalues.
The post on constrained ordination used dbrda to put environmental constraints on a Bray-Curtis dissimilarity. vegan has a second function for the same job, capscale, and most of the time the two are interchangeable. On a semimetric dissimilarity like Bray-Curtis they are not quite. They return the same constrained ordination, but a different pseudo-F, and the split happens at one specific place: the negative eigenvalues.
The two functions take different routes. capscale (Legendre and Anderson 1999) runs principal coordinates analysis on the dissimilarities, then ordinary RDA on the resulting coordinates. dbrda (McArdle and Anderson 2001) works directly on the double-centered distance matrix, without the intermediate coordinates. For a Euclidean distance the routes are equivalent. For a non-Euclidean one they diverge, because the dissimilarities cannot be embedded in real Euclidean space without an imaginary part, and that part shows up as negative eigenvalues.
A community and its dissimilarity
Two predictors drive composition; counts are Poisson around Gaussian niches. Bray-Curtis on this kind of data is semimetric, so it carries negative eigenvalues.
How non-Euclidean is this dissimilarity? Principal coordinates returns one eigenvalue per axis, and the negative ones are the imaginary part.
ev <- eigen(-0.5 * (diag(n) - 1/n) %*% as.matrix(D^2) %*% (diag(n) - 1/n),
symmetric = TRUE, only.values = TRUE)$values
c(positive = sum(ev > 1e-8), negative = sum(ev < -1e-8),
pct_negative = round(sum(abs(ev[ev < 0])) / sum(ev[ev > 0]) * 100, 1)) positive negative pct_negative
34.0 25.0 8.9 Many of the axes carry negative eigenvalues, and together they come to roughly nine percent of the positive inertia. Small, but it is exactly what the two functions handle differently.
The same axes
cap <- capscale(D ~ x1 + x2, data = env)
db <- dbrda(D ~ x1 + x2, data = env)
cap
Call: capscale(formula = D ~ x1 + x2, data = env)
Inertia Proportion Rank
Total 10.5194
RealTotal 11.5525 1.0000
Constrained 7.6692 0.6639 2
Unconstrained 3.8832 0.3361 34
Imaginary -1.0330
Inertia is squared Bray distance
Eigenvalues for constrained axes:
CAP1 CAP2
4.489 3.180
Eigenvalues for unconstrained axes:
MDS1 MDS2 MDS3 MDS4 MDS5 MDS6 MDS7 MDS8
0.8466 0.6682 0.2935 0.2225 0.1865 0.1839 0.1486 0.1219
(Showing 8 of 34 unconstrained eigenvalues)db
Call: dbrda(formula = D ~ x1 + x2, data = env)
Inertia Proportion Rank RealDims
Total 10.5194 1.0000
Constrained 7.6671 0.7288 2 2
Unconstrained 2.8523 0.2712 57 33
Inertia is squared Bray distance
Eigenvalues for constrained axes:
dbRDA1 dbRDA2
4.488 3.179
Eigenvalues for unconstrained axes:
MDS1 MDS2 MDS3 MDS4 MDS5 MDS6 MDS7 MDS8
0.8464 0.6680 0.2920 0.2224 0.1844 0.1770 0.1484 0.1201
(Showing 8 of 57 unconstrained eigenvalues)Read the two partition tables together. Total inertia is the same. The constrained inertia and its proportion are the same to three figures, near seventy-three percent. The constrained eigenvalues match: CAP1 and dbRDA1, CAP2 and dbRDA2, agree to the third decimal. As a constrained ordination the two results are the same picture.
The difference is in the bookkeeping below the constrained part. capscale reports an Imaginary line: it has set the negative-eigenvalue inertia aside, outside both the constrained and the real unconstrained totals. dbrda has no imaginary line; its unconstrained total already nets the negative eigenvalues against the positive ones, which is why its unconstrained inertia is smaller.
rbind(
capscale = c(adjR2 = RsquareAdj(cap)$adj.r.squared,
resid = cap$CA$tot.chi),
dbrda = c(adjR2 = RsquareAdj(db)$adj.r.squared,
resid = db$CA$tot.chi)) adjR2 resid
capscale 0.6520672 3.883228
dbrda 0.7193353 2.852350The adjusted R-squared values are effectively identical, near 0.72: both functions agree on how much the predictors explain. The residual inertia is where they split, because one keeps the negative part and the other does not.
A different denominator, a different F
The permutation test divides constrained variation by residual variation. Same numerator, different residual, different pseudo-F.
set.seed(1); ac <- anova(cap, permutations = 999)
set.seed(1); ad <- anova(db, permutations = 999)
c(capscale_F = round(ac$F[1], 1), dbrda_F = round(ad$F[1], 1),
capscale_p = ac$`Pr(>F)`[1], dbrda_p = ad$`Pr(>F)`[1])capscale_F dbrda_F capscale_p dbrda_p
56.300 76.600 0.001 0.001 The dbRDA pseudo-F comes out larger, near seventy-seven against fifty-six. The mechanism is direct: dbrda subtracts the negative-eigenvalue inertia from the residual, so its denominator is smaller, so its F is larger. Here both p values land at the permutation floor and the conclusion is the same, but in a borderline case the choice of function could move the p value across a threshold. When the test is the point, that matters.
This is the residual structure that drives the gap:
ca <- db$CA$eig
dfe <- data.frame(idx = seq_along(ca), eig = as.numeric(ca),
sign = ifelse(ca < 0, "negative", "positive"))
ggplot(dfe, aes(idx, eig, fill = sign)) +
geom_col(width = .85) +
geom_hline(yintercept = 0, color = pal$body, linewidth = .3) +
scale_fill_manual(values = c(positive = pal$forest, negative = pal$terra), name = NULL) +
labs(x = "Residual (unconstrained) axis", y = "Eigenvalue") + th()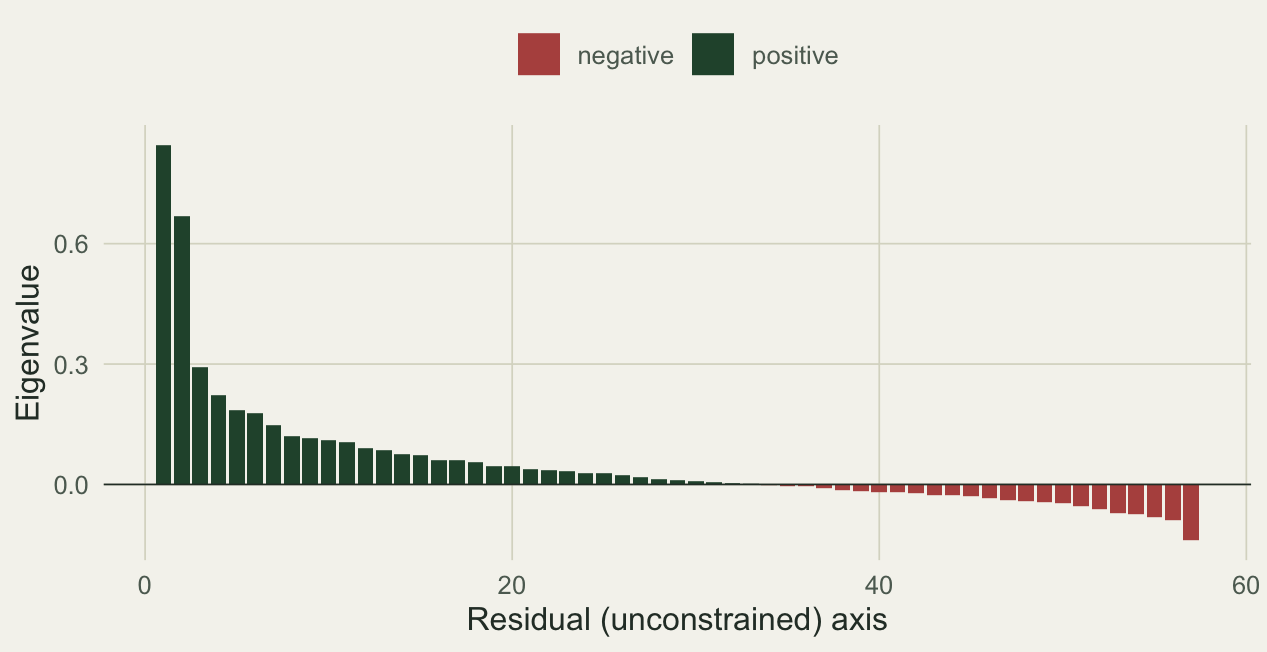
And the constrained ordination itself, which both functions agree on:
si <- as.data.frame(scores(db, display = "sites", choices = 1:2)); si$x1 <- x1
bp <- as.data.frame(scores(db, display = "bp", choices = 1:2))
mul <- 0.9 * max(abs(unlist(si[, 1:2]))) / max(sqrt(rowSums(bp^2)))
bp <- bp * mul
eig <- round(100 * db$CCA$eig / db$tot.chi, 1)
ggplot(si, aes(dbRDA1, dbRDA2, color = x1)) +
geom_point(size = 2.4) +
scale_color_gradient(low = pal$low, high = pal$high, name = "x1") +
geom_segment(data = bp, aes(0, 0, xend = dbRDA1, yend = dbRDA2), inherit.aes = FALSE,
arrow = arrow(length = unit(.18, "cm")), color = pal$terra, linewidth = .6) +
geom_text(data = bp, aes(dbRDA1, dbRDA2, label = rownames(bp)), inherit.aes = FALSE,
color = pal$terra, vjust = -.5, size = 3.5) +
labs(x = paste0("dbRDA1 (", eig[1], "%)"), y = paste0("dbRDA2 (", eig[2], "%)")) + th()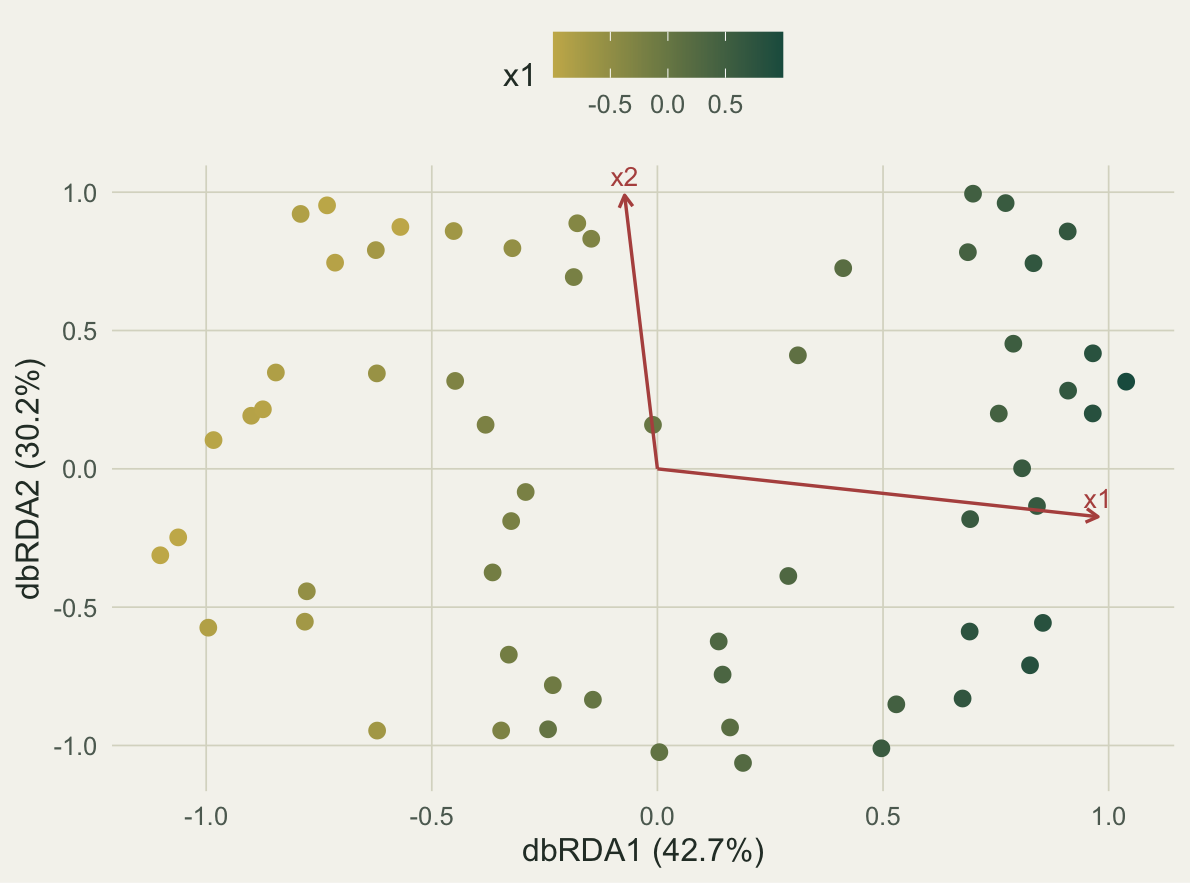
With a Euclidean distance they agree exactly
Hellinger distance is Euclidean, so it produces no negative eigenvalues and no imaginary part. With nothing to treat differently, the two functions return the same constrained inertia to numerical precision.
DH <- dist(decostand(comm, "hellinger"))
capH <- capscale(DH ~ x1 + x2, data = env)
dbH <- dbrda(DH ~ x1 + x2, data = env)
c(capscale = capH$CCA$tot.chi, dbrda = dbH$CCA$tot.chi,
difference = abs(capH$CCA$tot.chi - dbH$CCA$tot.chi)) capscale dbrda difference
1.518764e+01 1.518764e+01 1.598721e-14 The difference is at the rounding limit. The whole divergence on Bray-Curtis was the negative eigenvalues, and a Euclidean distance has none.
One practical difference, and what to use
capscale returns species scores by default, which is convenient for a biplot. dbrda does not, for the reason covered in the dbRDA post: the analysis runs on the distance matrix, not on a species table, so species coordinates are not produced unless you add them with sppscores.
sp_cap <- nrow(scores(cap, display = "species", choices = 1:2))
sp_db <- tryCatch(scores(db, display = "species", choices = 1:2), error = function(e) NULL)
list(capscale_species_scores = sp_cap,
dbrda_species_scores = if (is.null(sp_db) || all(is.na(sp_db))) "none" else nrow(sp_db))$capscale_species_scores
[1] 34
$dbrda_species_scores
[1] "none"The recommendation follows the mechanism. When the dissimilarity is metric or Euclidean (Hellinger, chord, Euclidean), the two functions agree, so use either; capscale is convenient if you want species scores for the biplot. When the dissimilarity is semimetric (Bray-Curtis, Jaccard) and the test is what you report, prefer dbrda: it partitions the variation directly from the distance matrix, including the non-Euclidean part, which is the partition McArdle and Anderson (2001) argued for. If you would rather stay with capscale on a semimetric dissimilarity, the negative eigenvalues can be removed first, by taking square roots of the dissimilarities or by adding a constant (add = TRUE); the vegan help documents both.
A note on the name. capscale takes its label from CAP, the canonical analysis of principal coordinates of Anderson and Willis (2003). That method is a discriminant or correlation analysis on principal coordinates, aimed at an a priori hypothesis, and it is not identical to what capscale computes, which is RDA on principal coordinates. The family is the same; the exact engine is not.
References
Legendre, P. and Anderson, M. J. (1999). Distance-based redundancy analysis: testing multispecies responses in multifactorial ecological experiments. Ecological Monographs 69(1): 1-24. https://doi.org/10.1890/0012-9615(1999)069[0001:DBRATM]2.0.CO;2
McArdle, B. H. and Anderson, M. J. (2001). Fitting multivariate models to community data: a comment on distance-based redundancy analysis. Ecology 82(1): 290-297. https://doi.org/10.1890/0012-9658(2001)082[0290:FMMTCD]2.0.CO;2
Anderson, M. J. and Willis, T. J. (2003). Canonical analysis of principal coordinates: a useful method of constrained ordination for ecology. Ecology 84(2): 511-525. https://doi.org/10.1890/0012-9658(2003)084[0511:CAOPCA]2.0.CO;2
Oksanen, J., Simpson, G. L., Blanchet, F. G., and others (2022). vegan: Community Ecology Package. R package version 2.6-4. https://CRAN.R-project.org/package=vegan